Schizophrenia is a severe psychiatric disorder affecting up to 1% of the worldwide population. Available therapy presents different limits comprising lack of efficiency in attenuating negative symptoms and cognitive deficits, typical features of schizophrenia and severe side effects. There is pressing requirement, therefore, to develop novel neuroleptics with higher efficacy and safety. Nitric oxide (NO), an intra- and inter-cellular messenger in the brain, appears to be implicated in the pathogenesis of schizophrenia. In particular, underproduction of this gaseous molecule is associated to this mental disease. The latter suggests that increment of nitrergic activity might be of utility for the medication of schizophrenia. Based on the above, molecules able to enhance NO production, as are NO donors, might represent a class of compounds candidates. Sodium nitroprusside (SNP) is a NO donor and is proposed as a promising novel compound for the treatment of schizophrenia.
- schizophrenia
- nitric oxide
- sodium nitroprusside
1. Schizophrenia
2. Nitric oxide (NO)
3. NO and Schizophrenia
4. Sodium Nitroprusside (SNP)
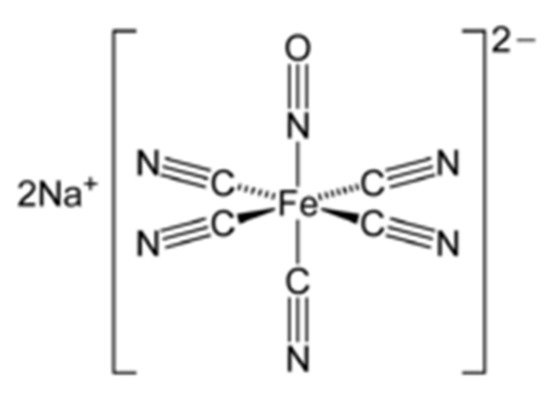
5. SNP and Schizophrenia
5.1. Preclinical Studies
| Species | Agent | Dose Range | Route | Behavioural Task | Effect | Reference |
|---|---|---|---|---|---|---|
| Rat | SNP | 0.3, 1, 3 mg/kg | i.p. acute | PPI | No effect | [45] |
| Rat | SNP | 2, 6 mg/kg | i.p. acute | Activity cage | Reversed PCP-induced hypermotility, stereotypies, ataxia | [46] |
| PCP | 5 mg/kg | i.p. acute | ||||
| Rat | SNP | 0.3, 1 mg/kg | i.p. acute | ORT | Reversed apomorphine-induced recognition memory deficits | [47] |
| Apomorphine | 1 mg/kg | i.p. acute | ||||
| Mouse | SNP | 2.5 mg/kg | i.p. acute | PPI | Reversed amphetamine-induced attentional deficits | [48] |
| Amphetamine | 10 mg/kg | i.p. acute | ||||
| Mouse | SNP | 4 mg/kg | i.p. acute | OFT | Prevented ketamine-induced hypermotility and stereotypies | [49] |
| Ketamine | 25 mg/kg | i.p. acute | ||||
| Rat | SNP | 5 mg/kg | i.p. acute | ORT | Impaired STM but counteracted ketamine-induced LTM deficits | [50] |
| Ketamine | 30 mg/kg | i.p. acute | OFT | Reversed ketamine-induced hypermotulity | ||
| Rat | SNP | 0.3, 1 mg/kg | i.p. acute | ORT | Reversed ketamine-induced recognition memory deficits | [51] |
| Ketamine | 3 mg/kg | i.p. acute | ||||
| SNP | 1 mg/kg | i.p. acute | SIT | Reversed ketamine-induced social isolation | ||
| Ketamine | 8 mg/kg | i.p. subchronic | ||||
| Rat | SNP | 2, 5 mg/kg | i.p. acute | TUNLT | Ineffective on MK-801-induced memory deficits. | [52] |
| MK-801 | 0.05, 0.1 mg/kg | i.p. acute | Minor effects on task accuracy and perseveration. | |||
| SHR Rat | SNP | 0.5, 2.5 mg/kg | i.p. chronic | Activity cage | Attenuated hypermotility in the SHR rat | [53] |
| SNP | 2.5 mg/kg | i.p. chronic | SIT | Attenuated social isolation in the SHR rat | ||
| SNP | 0.5, 1, 2.5 mg/kg | i.p. chronic | CFCT | Reversed memory deficits in the SHR rat | ||
| Mouse | SNP | 1 mg/kg | i.p. acute | PPI | Combination of subthresold doses of SNP and clozapine reversed amphetamine but no MK-801 induced attentional deficits | [54] |
| Clozapine | 1 mg/kg | i.p. acute | amphetamine but not MK-801-induced attentional deficits | |||
| Amphetamine | 5 mg/kg | i.p. acute | ||||
| MK-801 | 0.5 mg/kg | i.p. acute | ||||
| SNP | 2.5, 3.5, 4 mg/kg | i.p. acute | No effect | |||
| MK-801 | 0.5 mg/kg | i.p. acute | ||||
| Rat | SNP | 2.5 mg/kg | i.p. acute | OFT | Attenuated MK-801-induced hypermotility | [55] |
| MK-801 | 0.4 mg/kg | i.p. acute | PPI | No effect | ||
| Y-maze | No effect | |||||
| Rat | SNP | 2.5 mg/kg | i.p. acute | CART | [56] | |
| Risperidone | 0.25 mg/kg | i.p. acute | Combination of SNP and risperidone attenuated behavior avoidance behaviour |
5.2. Clinical Studies
| Design of Study | Evaluation | Participants | Agent | Dose Range | Route Outcome Measure | Effect | Reference |
|---|---|---|---|---|---|---|---|
| Double-blind placebo-controlled | Just after infusion | 20 patients (19–40 years old) |
SNP | 0.5 μg/min × 4 h | i.v. BPRS-18 PANSS |
Effective and safe | [58] |
| Double-blind placebo-controlled | Just after infusion | 18 patients | SNP | 0.5 μg/min × 4 h | i.v. Cognitive tests | Improvement of executive functions and safe | [59] |
| Double-blind placebo-controlled | Just after infusion/four weeks later | 20 patients (18–60 years old) |
SNP | 0.5 μg/min × 4 h | i.v. BPRS-18 PANSS CANTAB |
Ineffective but safe | [60] |
| Double-blind placebo-controlled | Just after the first and second infusion | 42 patients (18–45 years old) |
SNP | 0.5 μg/min × 4 h (twice at one week interval) | i.v. PANSS Cognitive tests |
Ineffective but safe | [61] |
| Double-blind placebo-controlled | Just after infusion/one week later | 52 patients (18–65 years old) |
SNP | 0.5 μg/min × 4 h | i.v. PANSS | Ineffective but safe | [62] |
| Double-blind placebo-controlled | Just after infusion 4 follow up evaluations |
20 treatment-resistant patients (18–60 years old) |
SNP | 0.5 μg/min × 4 h (four times) at 2 weeks of interval | i.v. PANSS BPRS-18 |
Ineffective and safe | [63] |
5.3. Potential Mechanism(s) of Action of SNP in Schizophrenia
| Normalization of the functionality of the NMDA-nNOS-cGMP pathway |
| Alleviation of cerebral hypoperfusion |
| Normalization of the functionality of the glutamatergic and dopaminergic neurotransmission |
| Potent antioxidant properties |
This entry is adapted from the peer-reviewed paper 10.3390/molecules26113196
References
- Freedman, R. Schizophrenia. N. Engl. J. Med. 2003, 349, 1738–1749.
- Lewis, D.A.; Lieberman, J.A. Catching up on schizophrenia: Natural history and neurobiology. Neuron 2000, 28, 325–334.
- Van Os, J.; Kenis, G.; Rutten, B.P. The environment and schizophrenia. Nature 2010, 468, 203–212.
- Weinberger, D.R. Implications of normal brain development for the pathogenesis of schizophrenia. Arch. Gen. Psychiatry 1987, 44, 660–669.
- Bitanihirwe, B.K.; Woo, T.U. Oxidative stress in schizophrenia: An integrated approach. Neurosci. Biobehav. Rev. 2011, 35, 878–893.
- Steeds, H.; Carhart-Harris, R.L.; Stone, J.M. Drug models of schizophrenia. Ther. Adv. Psychopharmacol. 2015, 5, 43–58.
- Javitt, D.C. Glutamate and schizophrenia: Phencyclidine, N-methyl-D-aspartate receptors, and dopamine-glutamate interactions. Int. Rev. Neurobiol. 2007, 78, 69–108.
- Pratt, J.; Winchester, C.; Dawson, N.; Morris, B. Advancing schizophrenia drug discovery: Optimizing rodent models to bridge the translational gap. Nat. Rev. Drug Discov. 2012, 11, 560–579.
- Lewis, D.A.; Pierri, J.; Volk, D.; Melchitzky, D.; Woo, T. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol. Psychiatry 1999, 46, 616–626.
- Field, J.R.; Walker, A.G.; Conn, P.J. Targeting glutamate synapses in schizophrenia. Trends Mol. Med. 2011, 17, 689–698.
- Abbott, A. Schizophrenia: The drug deadlook. Nature 2010, 468, 158–159.
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests a role as intercellular messenger in the brain. Nature 1988, 336, 385–387.
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258.
- Arnold, W.P.; Mittal, C.K.; Katsuki, S.; Murad, F. Nitric oxide activates guanylate cyclase and increases guanosine 3′5′-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. USA 1977, 74, 3203–3207.
- Kleppisch, T. Phosphodiesterases in the central nervous system. Hand. Exp. Pharmacol. 2009, 191, 71–92.
- Socco, S.; Bovee, R.H.; Palczewski, M.B.; Hickok, J.R.; Thomas, D.D. Epigenetics: The third pillar of nitric oxide signaling. Pharmacol. Res. 2017, 121, 52–58.
- Oh, S.J.; Fan, X. Current understanding on the role of nitric oxide and therapeutic potential of NO supplementation in schizophrenia. Schizophr. Res. 2020, 222, 23–30.
- Hibbs, J.B., Jr.; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric oxide a cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94.
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526.
- Prast, H.; Philippu, A. Nitric oxide as modulator of neuronal function. Prog. Neurobiol. 2001, 64, 51–68.
- Pitsikas, N. The role of nitric oxide in the object recognition memory. Behav. Brain Res. 2015, 285, 200–207.
- Lonart, G.; Wang, J.; Johnson, K.M. Nitric oxide induces neurotransmitter release from the hippocampal slices. Eur. J. Pharmacol. 1992, 220, 271–272.
- West., A.R.; Galloway, M.P.; Grace, A.A. Regulation of striatal dopamine neurotransmission by nitric oxide: Effector pathways and signalling mechanisms. Synapse 2002, 44, 227–245.
- Trabace, L.; Cassano, T.; Tucci, P.; Steardo, L.; Kendrick, K.M.; Cuomo, V. The effects of nitric oxide on striatal serotoninergic transmission involve multiple targets: An in vivo microdialysis study in the awake rat. Brain Res. 2004, 1008, 293–298.
- Calabrese, V.; Mancuso, C.; Calvani, M.; Rizzarelli, E.; Butterfield, D.A.; Stella, A.M. Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity. Nat. Rev. Neurosci. 2007, 8, 766–775.
- Bernstein, H.G.; Bogerts, B.; Keilhoff, G. The many faces of nitric oxide in schizophrenia. A review. Schizophr. Res. 2005, 78, 69–86.
- Bernstein, H.G.; Keilhoff, G.; Steiner, J.; Dobrowonly, H.; Bogerts, B. Nitric oxide and schizophrenia. Present knowledge and emerging concepts of therapy. CNS Neurol. Disord. Drug Targets 2011, 10, 792–807.
- Reif, A.; Herterich, S.; Strobel, A.; Ehlis, A.C.; Saur, D.; Jacob, C.P.; Wienker, T.; Topner, T.; Fritzen, S.; Walter, U.; et al. A neuronal nitric oxide (NOS-1) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol. Psychiatry 2006, 11, 286–300.
- Akbarian, S.; Bunney, W.E.; Potkin, S.G.; Wigal, S.B.; Hagman, J.O.; Sandman, C.A.; Jones, E.G. Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch. Gen. Psychiatry 1993, 50, 169–177.
- Lauer, M.; Johannes, S.; Fritzen, S.; Senitz, D.; Riederer, P.; Reif, A. Morphological abnormalities in nitric-oxide-synthase-positive striatal interneurons of schizophrenic patients. Neuropsychobiology 2005, 52, 111–117.
- Bernstein, H.G.; Stanarius, A.; Baumann, B.; Henning, H.; Krell, D.; Danos, P.; Falkai, P.; Bogerts, B. Nitric oxide synthase-containing neurons in the human hypothalamus: Reduced number of immunoreactive cells in the paraventricular nucleus of depressed patients and schizophrenics. Neuroscience 1998, 83, 867–875.
- Xing, G.; Chavko, M.; Zhang, L.X.; Yang, S.; Post, R.M. Decreased calcium-dependent constitutive nitric oxide synthase (eNOS) activity in prefrontal cortex in schizophrenia and depression. Schizophr. Res. 2002, 58, 21–30.
- Suzuki, E.; Nakaki, T.; Nakamura, M.; Miyaoka, H. Plasma nitrite levels in deficit versus non-deficit forms of schizophrenia. J. Psychiatry Neurosci. 2003, 28, 288–292.
- Lee, B.H.; Kim, Y.K. Reduced plasma nitric oxide metabolites before and after antipsychotic treatment in patients with schizophrenia compared to controls. Schizophr. Res. 2008, 104, 36–43.
- Nakano, Y.; Yoshimura, R.; Nakano, H.; Ikenouchi-Sugita, A.; Hori, H.; Umene-Nakano, W.; Ueda, N.; Nakamura, J. Association between plasma nitric oxide metabolites levels and negative symptoms of schizophrenia: A pilot study. Hum. Psychopharmacol. 2010, 20, 139–144.
- Das, I.; Khan, N.S.; Puri, B.K.; Hirsch, S.R. Elevated endogenous nitric oxide synthase inhibitor in schizophrenic plasma may reflect abnormalities in brain nitric oxide production. Neurosci. Lett. 1996, 215, 209–211.
- Das, I.; Ramchand, C.N.; Gliddon, A.; Hirsch, S.R. Nitric oxide, free radicals and polyamines may have a role in membrane pathology of schizophrenia. Neuropsychobiology 1998, 37, 65–67.
- Ramirez, J.; Garnica, R.; Boll, M.C.; Montes, S.; Rios, C. Low concentrations of nitrite and nitrate in the cerebrospinal fluid from schizophrenic patients. Schizophr. Res. 2004, 68, 357–361.
- Eastwood, S.L.; Harrison, P.J. Interstitial white matter neurons express less reelin and are abnormally distributed in schizophrenia: Towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol. Psychiatry 2003, 8, 821–831.
- Benes, F.M.; Berretta, S. GABAergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001, 25, 1–27.
- Connor, C.M.; Guo, Y.; Akbarian, S. Cingulate white matter neurons in schizophrenia and bipolar disorder. Biol. Psychiatry 2009, 66, 486–493.
- Miller, M.R.; Megson, I.L. Recent development in nitric oxide donor drugs. Br. J. Pharmacol. 2007, 151, 305–321.
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Giardina, P. Pharmacological modulation of nitric oxide release: New pharmacological perspectives, potential benefits and risks. Curr. Med. Chem. 2010, 17, 61–73.
- Godinez-Rubi, M.; Rojas-Mayorquin, A.E.; Ortuno-Sahagun, D. Nitric oxide donors as neuroprotective agents after an ischemic stroke-related inflammatory reaction. Oxid. Med. Cell. Longev. 2013, 97357.
- Wiley, J.L.; Golden, K.M.; Bowen, S.E. Effects of modulation of nitric oxide on acoustic startle responding and prepulse inhibition in rats. Eur. J. Pharmacol. 1997, 328, 125–130.
- Bujas-Bobanovic, M.; Bird, D.C.; Robertson, H.A.; Dursun, S.M. Blockade of phencyclidine-induced effects by a nitric oxide donor. Br. J. Pharmacol. 2000, 130, 1005–1012.
- Gourgiotis, I.; Kampouri, N.; Koulouri, V.; Lempesis, I.; Prasinou, M.; Georgiadou, G.; Pitsikas, N. Nitric oxide modulates apomorphine-induced recognition memory deficits in rats. Pharmacol. Biochem. Behav. 2012, 102, 507–514.
- Issy, A.C.; Pedrazzi, J.F.C.; Yoneyama, B.H.; Del Bel, E.A. Critical role of nitric oxide in the modulation of prepulse inhibition in Swiss mice. Psychopharmacology 2014, 231, 663–672.
- Maja-de-Oliveira, J.P.; Lobao-Soares, B.; Ramalho, T.; Gavioli, E.C.; Soares, V.P.; Teixeira, L.; Baker, G.B.; Dusun, M.S.; Hallak, J.C.E. Nitroprusside single-dose prevents the psychosis-like behavior induced by ketamine in rats up to one week. Schizophr. Res. 2015, 162, 211–215.
- Kandratavicius, L.; Balista, P.A.; Wolf, D.C.; Abrao, J.; Evora, P.R.; Rodriguez, A.J.; Chaves, C.; Maia-de-Oliveira, J.P.; Leite, J.P.; Dursun, S.M.; et al. Effects of the nitric oxide-related compounds in the acute ketamine animal model of schizophrenia. BMC Neurosci. 2015, 16, 9.
- Trevlopoulou, A.; Touzlatzi, N.; Pitsikas, N. The nitric oxide donor sodium nitroprusside attenuates recognition memory deficits and social withdrawal produced by the NMDA receptor antagonist ketamine and induces anxiolytic-like behaviour in rats. Psychopharmacology 2016, 233, 1045–1054.
- Hurtubise, J.L.; Marks, D.N.; Davies, D.A.; Catton, J.K.; Baker, G.B.; Howland, J.G. MK-801-induced impairments on the trial-unique, delayed nonmatching-to-location task in rats: Effects of acute sodium nitroprusside. Psychopharmacology 2017, 234, 211–222.
- Diana, M.C.; Peres, F.F.; Justi, V.; Bressan, R.A.; Lacerda, A.L.T.; Crippa, J.A.; Hallak, J.C.E.; Costhek-Abilio, V. Sodium nitroprusside is effective in preventing and/or reversing the development of schizophrenia-related behaviors in an animal model: The SHR strain. CNS Neurosci. Ther. 2018, 24, 624–632.
- Issy, A.C.; dos Santos-Pereira, M.; Cordeiro-Pedrazzi, J.F.; Cussa-Kubrusly, R.C.; Del-Bel, E.A. The role of striatum and prefrontal cortex in the prevention of amphetamine-induced schizophrenia-like effects mediated by nitric oxide compounds. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 353–362.
- Wang, X.; Ding, S.; Lu, Y.; Jiao, Z.; Zhang, L.; Zhang, Y.; Yang, Y.; Zhang, Y.; Li, D.; Lv, L. Effects of sodium nitroprusside in the acute dizolcipine (MK-801) animal model of schizophrenia. Brain Res. Bull. 2019, 147, 140–147.
- Titulaer, J.; Malmerfelt, A.; Marcus, M.M.; Svensson, T.H. Enhancement of the antipsychotic effect of risperidone by sodium nitroprusside in rats. Eur. Neuropsychopharmacol. 2019, 29, 1282–1287.
- Pitsikas, N. The role of nitric oxide synthase inhibitors in schizophrenia. Curr. Med. Chem. 2016, 23, 2692–2705.
- Hallak, J.C.E.; Maia-De-Oliveira, J.P.; Abrao, J.; Evora, P.R.; Zuardi, A.W.; Crippa, J.E.; Belmonte-de Abreu, P.; Baker, G.B.; Dursun, S.M. Rapid improvement of acute schizophrenia symptoms after intravenous sodium nitroprusside. A randomized, double-blind, placebo-controlled trial. JAMA Psychiatry 2013, 70, 668–676.
- Maja-de-Oliveira, J.P.; Abrao, J.; Evora, P.R.; Zuardi, A.W.; Crippa, J.A.; Belmonte-de-Abreu, P.; Baker, G.B.; Dursun, S.M.; Hallak, J.C.E. The effects of sodium nitroprusside treatment on cognitive deficits in schizophrenia: A pilot study. J. Clin. Psychopharmacol. 2015, 35, 83–85.
- Stone, J.M.; Morrison, P.D.; Koychev, I.; Gao, F.; Reilly, T.J.; Kolanko, M.; Mohammadinasab, M.; Kapur, S.; McGuire, P.K. The effect of sodium nitroprusside on psychotic symptoms and spatial working memory in patients with schizophrenia: A randomized, double-blind, placebo-controlled trial. Psychol. Med. 2016, 46, 3443–3450.
- Wang, X.; Zhao, J.; Hu, Y.; Jiao, Z.; Lu, Y.; Ding, M.; Kou, Y.; Li, B.; Meng, F.; Zhao, H.; et al. Sodium nitroprusside treatment for psychotic symptoms and cognitive deficits of schizophrenia. A randomized, double-blind, placebo-controlled trial. Psychiatry Res. 2018, 269, 271–277.
- Brown, H.E.; Freudenreich, O.; Fan, X.; O’ Heard, S.; Goff, D.; Petrides, G.; Harrington, A.L.; Kane, J.M.; Judge, H.; Hoeppner, B.; et al. Efficacy and tolerability of adjunctive intravenous sodium nitroprusside treatment for outpatients with schizophrenia: A randomized clinical trial. JAMA Psychiatry 2019, 76, 691–699.
- Adelino, M.P.M.; Nunes, M.V.; Nunes, M.F.Q.; Costa, E.R., Jr.; Ajub, E.; Mitrovich, M.P.B.; Ushirohira, J.M.; Quarantini, L.C.; Hallak, J.C.E.; Lacerda, A.L.T. Treatment-resistant schizophrenia-A RCT on the effectiveness of repeated-dose sodium nitroprusside. Schizophr. Res. 2021, 231, 70–72.
- Kocygit, Y.; Yoca, G.; Karahan, S.; Ayhan, Y.; Yazici, M.K. L arginine add-on treatment for schizophrenia: A randomized, double-blind, placebo controlled, crossover study. Turk. Psychiatri. Derg. 2018, 29, 147–153.
- Merritt, K.; Catalan, A.; Cowley, S.; Demjaha, A.; Taylor, M.; McGuire, P.; Cooper, R.; Morrison, P. Glyceryl trinitrate in first-episode psychosis unmedicated with antipsychotics: A randomized controlled pilot study. J. Psychopharmacol. 2020, 34, 839–847.
- Guimaraes, T.M.; Guimaraes, M.R.C.; Oliveira, I.A.F.; Leoni, R.F.; Santos, A.C.; Dursun, S.M.; Crippa, J.A.S.; Bressan, R.A.; Machado-de-Sousa, J.P.; Lacerda, A.L.T.; et al. Mononitrate isosorbide as an adjunctive therapy in schizophrenia. J. Clin. Psychopharmacol. 2021, 41, 260–266.
- Pinkham, A.; Loughead, J.; Ruparel, K.; Wu, W.C.; Overton, E.; Gur, R.; Gur, R. Resting quantitative cerebral blood flow in schizophrenia measured by pulsed arterial spin labeling perfusion MRI. Psychiatry Res. 2011, 194, 64–72.
- Keilhoff, G.; Becker, A.; Grecksch, G.; Wolf, G.; Bernstein, H.G. Repeated application of ketamine to rats induces changes in the hippocampal expression of parvalbumin, neuronal nitric oxide synthase and cFOS expression similar to those found in human schizophrenia. Neuroscience 2004, 126, 591–598.
- Xu, T.X.; Sotnikova, T.D.; Liang, C.; Zhang, J.; Jung, J.U.; Spealman, R.D.; Gainetdinov, R.R.; Yao, W.D. Hyperdopaminergic tone erodes prefrontal long-term potential via a D2 receptor operated protein phosphatase gate. J. Neurosci. 2009, 29, 14086–14099.
- Arroyo-Garcia, L.E.; Rodriguez-Moreno, A.; Flores, G. Apomorphine effects on hippocampus. Neural Regen. Res. 2018, 13, 2064–2066.
- Bohme, G.A.; Bon, C.; Stutzmann, J.M.; Doble, A.; Blanchard, J.C. Possible involvement of nitric oxide in long-term potentiation. Eur. J. Pharmacol. 1991, 199, 379–381.
- de Oliveira, L.; Spiazzi, C.M.; Bortolin, T.; Canever, L.; Petronilho, F.; Mina, F.G.; Dal Pizzol, F.; Quevedo, J.; Zugno, A.I. Different sub-anesthetic doses of ketamine increase oxidative stress in the brain of rats. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 1003–1008.
- Moreira, J.C.F.; Dal Pizzol, F.; Bonatto, F.; Gomez Da Silva, E.; Flores, D.G.; Picada, J.N.; Roesler, R.; Pegas Henriques, J.A. Oxidative damage in brains of mice treated with apomorphine and its oxidized derivative. Brain Res. 2003, 992, 246–251.