Hypoxia-driven metabolic adaptation of cancer cells revolutionizes the current understanding of hypoxia-associated tumor development and progression. Several hypoxia-responsive molecular determinants, such as hypoxia-inducible factors, guide the cellular adaptation to hypoxia by gene activation, which is critical for promoting malignant progression in the hostile tumor microenvironment. This article fully recaps the major research progress in this topic in order to assess its potential as a targetable pathway to better combat cancer and overcome treatment resistance. Knowledge from this article facilitates cancer researchers to sharpen their focus on gaining mechanistic insights into metabolic transformation in hypoxic cancers and developing novel strategies to target tumor metabolic rewiring driven by hypoxia, which would open avenues to improve the management of tumor hypoxia.
- hypoxia
- metabolic reprogramming
- tumor microenvironment
- progression and metastasis
- Introduction
Oxygen is indispensable for cellular metabolism in multicellular organisms. A set of metabolic reactions involving oxidative phosphorylation (OXPHOS) utilizes the oxygen to generate adenosine triphosphate (ATP), which is the molecular currency in the intracellular energy transfer and essential to develop almost all cellular and biological processes. No doubt, cells are provisioned with specialized chemoreceptors to sense increased (hyperoxia) or decreased (hypoxia) oxygen levels [1]. As a result, adaptive processes are stimulated during physiological conditions that compromise the oxygen environment (e.g., embryonic development and exercise), permitting tight control of the homeostasis [2,3]. Otherwise, decompensation in oxygen regulation may lead to the development and/or progression of several pathologic disorders, including inflammation, cardiovascular diseases, and cancer [4].
Tumor cells are mainly characterized by their ability to sustain chronic proliferation. The fast-paced growth demands modifications in the tumor metabolism to support the high rate of energy requirements [5]. Furthermore, the newly formed and dysfunctional vascular supply produces areas of tumor hypoxia that demand alternative metabolic pathways nondependent of oxygen [4]. The reprogramming energy metabolism by tumor cells has been recognized and well-characterized as one of the emerging hallmarks of cancer [5]. Nevertheless, the effect of hypoxia in the adjustment of cancer metabolism has recently gained significant attention, not only for the hypoxic areas that cause the prompt growing tumor but also for the hypoxic effect in the tumor microenvironment that produces the antiangiogenic factors [6]. In order to develop new therapeutic strategies that improve the efficacy of antineoplastic agents and overcome the treatment resistance, we take a detailed look at the biological processes, key events, and consequences regarding the hypoxia-driven metabolic adaptation of tumor cells.
- Biologic Aspects of Tumor Hypoxia
Tumor hypoxia results from decreased microenvironment oxygen availability. Under physiological conditions, the average oxygen tension in normal tissues is approximately 40 mmHg [7]. The parameters to recognize the reduced oxygen levels may vary widely among several types of cancer. A general approach for solid tumors considers the tumor hypoxic thresholds in the range of 0.02 mmHg to 35 mmHg [8]. Tumor cells adapt to hypoxic conditions, inducing genomics, and proteomics modifications that lead to structural and functional alterations. A decreased proliferation rate is the main physiologic feature that distinct hypoxic from normoxic cells. Cells under hypoxic conditions switch to a quiescent, apoptotic, or even necrotic status [4]. Moreover, recent in vitro models recreating a microenvironment with oxygen restriction provide evidence that hypoxia can promote the expression of stem-cell-associated genes, inducing cancer stem cell properties [9]. Remarkably, the interruption of the proliferative signaling is compensated by the continuous induction of angiogenesis regulated mainly by the expression of vascular endothelial growth factor-A (VEGF-A) [10]. Thus, hypoxic niches generate a heterogeneity population of malignant cells that contribute to the aggressiveness of progression and spread.
- Genetic and Metabolic Modifications in the Hypoxic Tumor Microenvironment
Tumor hypoxia through HIF-1α regulation drives genetic instability in a large group of genes that determine malignant and aggressive molecular features. Failures in DNA damage repair alter cell cycle checkpoints and chromosomal aberrations, leading to genomic changes related to deficiencies in oxygenation [39]. In turn, numerous genes that encode metabolites and enzymes involved in metabolic processes contribute to tumor metabolic reprogramming.
Oxygen is an indispensable factor for aerobic metabolism. After glucose breakdown, the pyruvate into the tricarboxylic acid cycle (TCA), and then the oxygen is used as a terminal electron acceptor in the OXPHOS to produce ATP. Besides ATP, the set of metabolic reactions generates carbon dioxide and a molecule of water. Adaptive responses to deficiencies in oxygenation allow the cells to significantly reduce the amount of pyruvate, which transfers to the mitochondria and switches from OXPHOS to substrate phosphorylation to guarantee the ATP production. The metabolic reaction generates lactic acid as a waste product instead of carbon dioxide [5]. The alternative metabolic pathways confer tumor cells to survive and grow in the microenvironment with oxygen deprivation. Interestingly, tumor cells prefer substrate phosphorylation over OXPHOS even in the conditions with normal levels of oxygen due to this pathway providing cancer cells with important advantages. This process is so-called Warburg effect, known as aerobic glycolysis [40]. Despite aerobic metabolism being more productive generating 32 molecules ATP/mol glucose versus two molecules ATP/mol glucose createds through OXPHOS, increased glycolysis flux produces higher amounts of ATP in a shorter time frame [47]. It supplies the high demands of energy during tumorigenesis, tumor progression, and metastasis.
The metabolic intermediates produced in the glycolysis are used in the macromolecular biosynthesis of proteins, lipids, and amino acids, contributing not only to sustain the high rate of cell proliferation but also to the clearance of metabolic waste products. Lactate recycling occurs in surrounding normoxic cells which express monocarboxylate transporter 1 (MCT1) to import lactate and convert it to pyruvate for the mitochondrial metabolism [48]. Fatty acids are other essential substrates of tumor cells as they are the major component of cell membranes, facilitating the membrane fluidity, and maintaining mitochondrial activity. The mitochondrial aerobic process of breaking down a fatty acid into acetyl-CoA units is so-called fatty acid oxidation, which produces energy to fuel the cell [49]. Hypoxia, via the transcriptional machinery of HIF-1α, modulates the reprogramming of lipid metabolism, resulting in the support of fast tumor proliferation. In order to enhance lipogenesis, HIF-1α induces the transcription of genes involved in fatty acid uptake (e.g., PPARγ, FABP3, FABP4, and FABP7), endocytosis of lipoproteins (e.g., LRP1 and VLDLR), and fatty acids synthesis (e.g., SREBP-1 and FASN). Moreover, HIF-1α activates target genes involved in lipid accumulation (e.g., PLIN2 and HIG2) and inhibition of lipolysis (e.g., ATGL), once fatty acids are converted into neutral triacylglycerols (e.g., AGPAT2 and LIPIN-1) [50].
3.1. Promotion of Anaerobic Glycolysis
Facilitative glucose transporters (GLUTs) are a family of 14 membrane proteins responsible for extracellular glucose import. Tumor cells require a high amount of glucose, not only for the prompt rate of proliferation but also for the low efficiency of aerobic glycolysis. HIF-1α binds to cis-acting binding sites of the GLUT1 and GLUT3 genes inducing the overexpression, and as a result, the rate of import glucose is increased [51,52].
HIF-1α promotes the expression of crucial glycolytic enzymes hexokinase 2 (HK2) and phosphofructokinase 1 (PFK1), producing the acceleration of the glycolytic flux at different levels. HK2 phosphorylates the intracellular glucose to glucose 6-phosphate (G6P) into the investment phase [53]. PFK1 catalyzes the important control point of converting fructose 6-phosphate (F6P) to fructose 1,6-bisphosphate (FDP) and ADP, and it is regulated allosterically [54,55]. The promoter activities of PFK1 are upregulated through direct binding of HIF-1α in the gene promoter region [56]. Interestingly, PFK1 is encoded by the gene 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 (PFKFB2) that is also induced by HIF-1α. [57]. Another direct target gene of HIF-1α is lactic dehydrogenase A (LDHA). HIF-1α interacts with the HRE-D site in the LDHA gene promoter to induce its expression [58]. LDHA then participates in the last step of glycolysis, catalyzing pyruvate into lactate with concomitant generation of nicotinamide adenine dinucleotide (NAD+) from NADH [59].
As a consequence of the increased rate of aerobic glycolysis, the lactate synthesis rises and HIF-1α allows to regulate the intracellular acidification through the expression of the transmembrane protein MCT4, aimed to transport lactate to the extracellular environment [60]. The extracellular lactate coupled with the overexpression of carbonic anhydrase IX and XII (CA-IX and CA-XII) regulated by HIF-1α, promote the acidification of the tumor microenvironment ranging from 6.0 to 6.5. It is important to recognize that the acidic microenvironment under oxygen availability may also upregulate CA-IX independent of the HIF-1α pathway, though sharing the same transcriptional factors induced by HIF-1α [61] (Figure 1A).
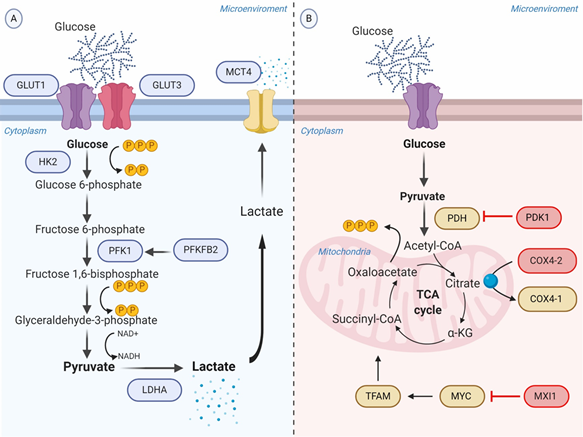
Figure 1. Schematic representation of the glycolytic switch mediated by tumor hypoxia. (A) Regulation of genes encoding glycolytic enzymes involved in the promotion of anaerobic glycolysis. GLUT1 and GLUT3 overexpression increase the rate of glucose import. HK2 phosphorylates the intracellular glucose to glucose 6-phosphate. PFK1, produced by PFKFB2 or induced by HIF-1α, converts Fructose 6-phosphate into Fructose 1,6-bisphosphate and ADP. LDHA catalyzes pyruvate into lactate with concomitant generation of NAD+ from NADH. MCT4 transports lactate to the extracellular environment. (B) Repression of OXPHOS. PDK1 inactivates PDH and blocks the conversion of pyruvate to acetyl-CoA. MXI1 inhibits the transcriptional activity of MYC and thereby inactivates MYC target genes (e.g., TFAM) involved in OXPHOS and mitochondrial dynamics, replication, and biogenesis. The generation and accumulation of ROS are controlled by the activation of the mitochondrial proteases COX4-2, responsible for the degradation of the COX4-1 isoform. Abbreviations: ADP, adenosine diphosphate; COX, cytochrome c oxidase; GLUTs, facilitative glucose transporters; HK2, hexokinase 2; LDHA, lactic dehydrogenase A; MCT, monocarboxylate transporter; MXI, MAX interactor; NAD+, nicotinamide adenine dinucleotide; OXPHOS, oxidative phosphorylation; PDH, pyruvate dehydrogenase; PDK1, Pyruvate dehydrogenase kinase; PFK1, phosphofructokinase-1; PFKFB2, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 2; ROS, reactive oxygen species; TFAM, transcription factor A, mitochondrial.
3.2. Repression of Oxidative Phosphorylation
Pyruvate dehydrogenase (PDH) is the mitochondrial enzyme responsible for the conversion of pyruvate to acetyl-CoA to consequent entry into the TCA cycle. HIF-1α induces the expression of pyruvate dehydrogenase kinase 1 (PDK1) gene encoding a kinase enzyme, which phosphorylates and inactivates PDH. Therefore, it represses OXPHOS and prevents aerobic respiration [62].
On the other hand, HIF-1α has the potential to inhibit mitochondrial metabolism through binding and transactivating the MAX interactor-1 (MXI1) gene. The protein encoded by this gene competes for MAX binding and inhibits the transcriptional activity of c-MYC function, and thereby inactivating c-MYC target genes involved in OXPHOS and mitochondrial dynamics, replication, and biogenesis [63]. For example, MXI1 can downregulate transcription factor A, mitochondrial (TFAM), which is essential for transcription, replication, and packaging of mtDNA into nucleoids [64]. Moreover, HIF-1α controls the mitochondrial function in hypoxic conditions via activating the mitochondrial proteases cytochrome oxidase 4-2 (COX4-2), which degrades the COX4-1 isoform to protects the cell from the harmful accumulation of ROS [65] (Figure 1B).
- Impact of Metabolic Reprogramming Driven-Hypoxia on Tumor Progression
Research in vitro and in vivo assessing tumor metabolic reprogramming driven by hypoxia continues to bring surprises to reveal not only its influence in the cell survival and tumorigenesis, but also in the tumor progression through immune escape, angiogenesis, metastasis, and radiotherapy and chemotherapy resistance.
Hypoxia-driven immunometabolic alternations have recently gained significant attention due to its capacity to downregulate the antitumor immune activity in various ways [66]. HIF-1α upregulates the transcription of genes encoding key glycolytic enzymes such as aldolase A (ALDA), phosphoglycerate kinase 1 (PGK1), and pyruvate kinase M (PKM), stimulating the glycolytic metabolism in immune cell populations [67]. In the first instance, it can promote the differentiation of highly glycolytic cells. However, the increased flux of glucose intake by tumor cells decreases the availability of extracellular glucose, which is required for immune cells for the ATP production and the synthesis of lipids, amino acids, and nucleotides [68]. Thus, glucose deficiencies may lead to immune cell dysfunction. High concentrations of glycolytic metabolites and the expression of cytokines and chemokines (e.g., CCL5, CXCL12, and CXCR4) driven by HIF-1α also generate an unfavorable scenario for immune cell recruitment. Indeed, an oxygen-deprived tumor microenvironment influences the migration and activity of both innate and adaptive immune cells [69].
The functions of dendritic cells (DCs) involve the presentation of tumor-derived antigens to naïve T cells to initiate the immune adaptive response. Hypoxia does not hamper DC maturation; however, in vitro assays support its important role in DC dysfunction through interfering with DC differentiation, adaptation, and activation. Hypoxia restricts the ability of immature DCs to capture antigens through the downexpression of Rho GTPase and ERM proteins. Moreover, DCs in response to low oxygen levels alter the expression of proinflammatory and proangiogenic cytokines [70].
The cascade of molecular events triggered by HIF-1α activation involves the secretion of cytokine and chemokines that attract monocytes, allowing for the differentiation into macrophages or tumor-associated macrophages (TAMs). TAMs represent the major inflammatory component of the tumor microenvironment and generate mechanisms to support the immune escape, such as the expression of the ligand-receptor PD-L1 that inhibits T cells cytotoxic function [71]. In addition, the concurrent inflammation induced by macrophages contributes to supporting the hallmark capabilities of tumor cells [5].
The acidic tumor microenvironment and the production of free oxygen radicals further reduce the migration and activity of polymorphonuclear leukocytes and cytotoxic T lymphocytes (CTLs). CTLs play a critical role in adaptive immune response promoting apoptosis in recognized tumor cells. Lack of CTL activity results in an immunosuppressive tumor microenvironment. Otherwise, hypoxia stimulates the recruitment of the immunosuppressive T cells regulatory (Tregs) through CCL28-CCR10, suppressing T-effector cells, and secreting VEGFA, which stimulate the tumor immune evasion [72] (Figure 2A).
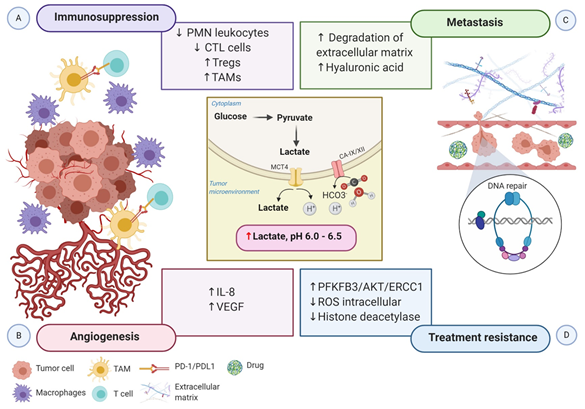
Figure 2. Schematic representation of the impact of hypoxia-associated metabolic reprogramming in tumor progression. The increased production of lactate along with the overexpression of CA-IX and CA-XII are regulated by HIF-1α directly, promoting the acidification of the tumor microenvironment. (A) The acidic microenvironment decreases the proliferation of polymorphonuclear leukocytes and CTL cells and increases the recruitment of Tregs cells. Moreover, the secretion of cytokines and chemokines attracts monocytes, enabling them to differentiate into macrophages or TAMs. TAMs express ligand-receptor PD-L1 that inhibits T cells cytotoxic function, and macrophages induce inflammation. Altogether, these mechanisms generate an immunosuppressive tumor microenvironment. (B) Elevated lactate in the tumor microenvironment boosts IL-8 and VEGF to facilitate angiogenesis. (C) Increased angiogenesis coupled with the acidic tumor microenvironment induces the degradation of the extracellular matrix and the expression of hyaluronic acid, which supports tumor cell migration and metastasis. (D) Lactate efflux activates metabolic substrates and the transcription of genes involved in metabolic rewiring to influence the tumor’s ability to respond to radiotherapy and chemotherapy-induced DNA damage. Increased PFKFB3 improves the ability of DNA repair, and reduced intracellular ROS accumulation increases the tolerance to DNA damage. Moreover, inactivation of histone deacetylase caused by elevated lactate promotes DNA repair. Abbreviations: CA, carbonic anhydrase; CTL, cytotoxic T lymphocytes; IL-8, interleukin 8; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand1; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; PMN, polymorphonuclear leukocytes; ROS, reactive oxygen species; TAMs, tumor-associated macrophages; Tregs, regulatory T cells; VEGF, vascular endothelial growth factor.
The hypoxia and HIF-1α pathways have been shown to regulate angiogenesis through the transcription of genes involved in angiogenesis, as well as factors antiangiogenic. Human endocrine gland derived VEGF, endoglin (ENG), leptin (LEP), low-density lipoprotein receptor-related protein 1 (LRP1), and TGF-β3 are the HIF-1α target genes that are implicated in different steps in angiogenesis [73].
Growing evidence indicates that tumor metabolic reprogramming driven by hypoxia also participates in angiogenesis via HIF-1α independent pathway. Elevated lactate accumulation into the extracellular matrix (ECM) resulted from the accelerated aerobic glycolysis promotes the accumulation of two potent promoters of angiogenesis, interleukin 8 (IL-8) and VEGF/VEGFR2 signaling pathway [74]. MCT1 increases the lactate influx from the ECM to inside of endothelial cells, triggering IL-8 expressions through the NF-ĸB signaling pathway, in a ROS and IĸBα-dependent manner [75]. Moreover, lactate stimulates tyrosine phosphorylation of the receptors tyrosine kinases Axl, Tie2, VEGFR-2, and ephrin type-A receptor 2 (EphA2), leading to the stimulation of the PI3K/Akt pathway in endothelial cells. Altogether, high levels of lactate potentiate the proliferation of endothelial cells, modulate the endothelial phenotype, and the neovessel formation, supplying the tumor with a supportive microenvironment [76] (Figure 2B).
Notably, HIF-1 participates in every critical aspect of cancer biology. While increased glycolytic metabolism empowers cancer cells to survive and evade immune attack, the molecular regulation of the epithelial to mesenchymal transition (EMT) and the new blood vessels confer malignant cells to invade and disseminate from the primary tumor to distant sites. Metabolic switching of tumor cells under hypoxic conditions, characterized by microenvironment acidification, also contributes to supporting tumor cell migration and metastasis. Lactic acid increases cancer cell motility, induces the synthesis of hyaluronic acid (HA) and the degradation of the ECM, and acts as a “natural selection regulator” that selective the most malignant phenotype profile of tumor cells [77,78] (Figure 2C).
Much of the interest in tumor metabolic network reprogram derives from its clinical impact on treatment resistance. The molecular mechanisms in the accelerated aerobic glycolysis and lactate efflux involve the transcription of genes, metabolic substrates, and events that influence the tumor responses to radiotherapy and chemotherapy at different levels. First, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), a key gene of aerobic glycolysis, improves the ability of DNA repair through the PFKFB3/AKT/ERCC1 signaling pathway [79]. Second, the repression of OXPHOS leads to the accumulation of intracellular ROS and increasing the tolerance to DNA damage. At last, lactate inactivates the class I and II histone deacetylases (HDAC), resulting in the hyperacetylation of histones H3 and H4, alteration of chromatin compactness, promotion of DNA repair gene upregulation, and initiation of the nuclear activation of DNA-dependent protein kinase, catalytic subunit (DNA-PKcs) [80]. In addition, experimental evidence in tumor xenografts demonstrated that elevated lactate levels are correlated with a decreased response to fractionated irradiation, perhaps due to the antioxidative capacity of lactate [81] (Figure 2D).
HIF-1α and HIF-2α are responsible for the transcriptional activity of some genes involved in the maintenance and evolution of cancer stem cells (CSCs), such as POU domain, class 5, transcription factor 1 (POU5F1) [82], delta-like 1 Homologue (DLK1) [83], CD133 [84], and CD24 [85]. Despite the low prevalence of CSCs (1–3%), this subpopulation has a high resistance to conventional therapy due to its ability to self-renewal, differentiation, and tumorigenicity. Recent evidence indicates that hypoxic niches modify the CSC and promote the EMT-like phenotype under the deregulation of phosphatase and tensin homolog (PTEN) [86]. Interestingly the metabolic switch induced for hypoxia also contributes to it regulating certain genes related to CSCs, through increased secretion of exosomes [87].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21155487