Despite the improvements in diagnostic and therapeutic approaches, breast cancer still remains one of the world’s leading causes of death among women. Particularly, triple negative breast cancer (TNBC) is characterized by aggressiveness, metastatic spreading, drug resistance and a very high percentage of death in patients. Nowadays, identification of new targets in TNBC appears very compelling. TNBC are considered negative for the estrogen receptor alpha (ERα) expression. Nevertheless, they often express ERβ and its variants. As such, this TNBC subtype still responds to estrogens. While the ERβ1 variant seems to act as a tumor-suppressor, the two variants ERβ2 and 5 exhibit pro-oncogenic activities in TNBC. Thus, ERβ1 activation might be used to limit the growth and spreading as well as to increase the drug sensitivity of TNBC. In contrast, the pro-oncogenic properties of ERβ2 and ERβ5 suggest the possible development and clinical use of specific antagonists in TNBC treatment. Furthermore, the role of ERβ might be regarded in the context of the androgen receptor (AR) expression, which represents another key marker in TNBC. The relationship between AR and ERβ as well as the ability to modulate the receptor-mediated effects through agonists/antagonists represent a challenge to develop more appropriate therapies in clinical management of TNBC patients.
1. Introduction
Breast cancer (BC) represents the second most diagnosed malignancy and the fifth commonest cause of cancer-related death worldwide [
1]. The BC incidence is higher in economically developed countries, probably because the disease’s onset is linked to risk factors, such as obesity, sedentary lifestyle, smoking, alcohol drinking, high consumption of red meat reach in hormones, use of oral contraceptives. Additionally, BC mortality is higher in countries with a low Human Development Index (HDI) [
2,
3].
Despite the advancements in early detection and treatment, BC often shows drug-resistance, likely due to its wide heterogeneity. BC is, indeed, characterized by different molecular signatures responsible for the disparate response to therapeutics and differences in patients’ long-term survival [
4]. To date, on the basis of the expression of the classical BC markers, estrogen receptor alpha (ERα), progesterone receptor (PR) and the human epidermal growth factor receptor II (Her2), the molecular classification divides BC in five subtypes. The luminal A and luminal B, which are both characterized by ERα expression, while differing from each other in Her2 over-expression in luminal B; the Her2-enriched subtype; the basal like subtype, lacking the expression of ERα and PR and the amplification of the Her2 gene, ERBB2; the normal like subtype, with molecular characteristics similar to normal breast epithelium. Among them, the basal like subtype is also known as triple-negative breast cancer (TNBC), lacking expression of the three most important BC markers [
5]. TNBC are currently treated with systemic chemotherapy. However, patients have a poor prognosis, poor recurrence-free and overall survival outcomes [
4,
5,
6]. These considerations highlight the need for new molecular targets in TNBC.
Estrogen receptor β (ERβ) is a sex steroid receptor and a transcription factor expressed in different cancers, such as prostate [
7,
8], colon [
9] and breast [
10]. ERβ has been detected in 30% of BC patients [
11]. As such, many reports in literature aim to determine the prognostic role of ERβ in TNBC, with conflicting results, likely because of the lack of specific antibodies and immuno-histochemistry (IHC) approaches [
12].
Nowadays, it is accepted that TNBC cells express different ERβ variants. ERβ1, which contains the entire predicted sequence, seems to play an inhibitory effect on TNBC growth and metastasis, while the truncated variants ERβ2 and 5 trigger proliferation and migration of TNBC. Therefore, the different ERβ variants represent promising molecular targets to develop precision strategies in TNBC clinical approaches [
13].
2. Genomic Action of ERβ in TNBC
The discovery of ERβ was enthusiastically received by endocrinologists and oncologists, since it suggested that the pleiotropic effects of estrogen can be mediated through ERβ and its isoforms, other than the well-known ERα. Recent advances in molecular analysis of pathways activated by ERs have allowed for identification of EREs in the promoters of numerous genes and to characterize changes in the gene expression profile upon estradiol treatment of TNBC cells. Since these TNBC cells do not express ERα, these findings have suggested that a different isoform of ER is present and functionally active in TNBC. Consistent with these hypotheses, many findings have reported an onco-suppressor role for ERβ in TNBC. Inducible expression of full-length ERβ in MDA-MB-468 cells and treatment with estradiol or the selective ERβ ligand, ERB-041, induces a G1 cell-cycle arrest, blocks the colony formation and reduces the tumor size in xenografted mice. The antagonists, ICI 182,780 or 4-hydroxy-tamoxifen, restore cell growth. RNA sequencing showed that most (about 80%) of the target genes regulated by ERβ are ligand-dependent, while only 20% are ligand-independent. The ligand-mediated growth-inhibitory effects of ERβ are due to regulation of target genes involved in the Wnt/β-catenin pathway and G1/S cell cycle checkpoint control, two critical steps in cancer cell proliferation [
36]. Notably, ERβ expression and activation by estradiol upregulates both the gene encoding the cyclin dependent kinase inhibitor p21 and CDKN1A, as well as the noncanonical Wnt ligand, WNT4, and the β-catenin interacting protein, CDH1. Downregulation of the Wnt inhibitor, DKK1, can also be observed [
36]. Consistent with these studies, the ligand-mediated ERβ activation by estrogens or the ERβ selective agonist, LY500307, decreases cell proliferation and blocks the cell cycle in doxycycline (Dox)-inducible ERβ expressing MDA-MB-231 cells. Microarray data and qPCR analysis showed that activation of ERβ suppresses the cell cycle-related genes, such as cyclin-dependent kinase 1 (CDK1), cyclin B and cyclin H [
37]. These findings are consistent with previous studies indicating that the ligand-mediated activation of ERβ suppresses proliferation in other cancer cell lines [
36,
37,
38].
An antimetastatic role for ERβ has also been proposed in TNBC. Treatment with estrogen or LY500307 induces changes in gene expression profiles of ERβ-expressing TNBC cells, with several (almost 976) differently regulated genes. Among them, some genes coding for interleukins and other inflammation-related factors are significantly inhibited by estrogen. In contrast, four members of a superfamily of cystatins (cystatins 1, 2, 4 and 5) are upregulated by the ligand. Because of the inhibition of the TGF-β/SMAD pathway, high cystatin 1, 2, 4 and 5 expression levels have been associated with improved relapse-free survival and decreased metastatic potential in TNBC patients [
39]. Consistent with this study, ERβ knockdown leads to the improper activation of TGF-β signalling pathway in TNBC models, thereby inducing the transcription of genes involved with either assembly, organization or in combination, of the extracellular matrix as well as the migration/invasion potential. ERβ agonists, such as ERB-041, WAY2000070, 3βA-diol and liquiritigenin, result in a significant decrease of TNBC cell invasiveness [
40]. By repressing epidermal growth factor receptor (EGFR) transcription, ERβ and its ligand 3βA-diol suppress insulin-like growth factor II (IGF-II) mRNA binding protein 3 (IMP3) in MDA-MB-231 and MDA-MB-468 cells. In this way, the receptor likely inhibits cell invasiveness. The specific ERβ antagonist, PHTPP (4-[2-Phenyl-5,7-bis(trifluoromethyl) pyrazolo [1,5-a] pyrimidin-3-y1] phenol), restores IMP3 and EGFR expression [
41]. A novel mechanism through which ERβ1 might inhibit invasiveness of TNBC cells has also been described. Most TNBC cells harbor a mutant version of p53 showing oncogenic functions, including the ability to promote metastasis. In MDA-MB-231 cells, ERβ upregulates SHARP-1 and CCNG2, which inhibit the metastatic events, while downregulating the pro-metastatic factor, follistatin. Thus, ERβ counteracts the oncogenic functions mediated by a p53 mutant and exerts its antimetastatic properties through a transcriptional mechanism [
42]. Further, ERβ1 inhibits migration and invasiveness of MDA-MB-231 and Hs578T cells by regulating the expression level of epithelial-mesenchymal transition (EMT) markers. The ability of ERβ to reduce tumor metastases has been further corroborated by findings showing that hyperexpression of ERβ1 in MDA MB231 and Hs578T cells induces a low recurrence of lung metastases in xenografted mice [
43].
Finally, given the presence of ERβ variants in target cells [
44], many reports have investigated their specific role in TNBC cells. MDA-MB-468 and BT-549 cells, for instance, express very low levels of ERβ1, which exerts antioncogenic properties. In contrast, the most abundant isoforms, ERβ2 and ERβ5, exhibit pro-oncogenic activities by acting on cell proliferation, migration and invasion [
13]. Thus, it might be argued that the final outcome of ERβ activation depends on expression and content of the receptor variants in TNBC.
Nowadays, it is largely recognized that genomic effects mediated by ERβ exert a potential anti-oncogenic role in TNBC. Specific targeting of ERβ might represent an interesting pharmacological option in TNBC patients who often exhibit or develop drug-resistance.
3. Non-Genomic Actions of ERβ in TNBC
Aside from the aforementioned genomic actions, cytoplasmic ERβ can also work in a non-genomic way upon ligand binding. These actions are much faster than the genomic ones, taking place in cytoplasm within seconds to minutes, and involving generation of second messengers, such as calcium [
45] and cAMP [
46] as well as activation of the MAPK pathway [
47] or interaction with components of the proteasome degradation pathway [
48]. These actions escape the transcription and translation inhibition.
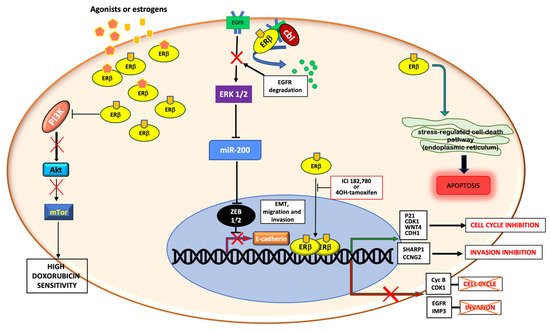
The PI3K/AKT pathway is crucial for growth, proliferation, angiogenesis and migration of BC [
49,
50] and is upregulated in aggressive TNBCs [
51]. A loss of function of the PI3-K inhibitor, PTEN, is often associated with a worse prognosis and outcome for BC patients. In TNBCs, increased ERβ1 expression correlates with downregulation of pAKT, which represents a favorable prognostic marker for the overall and disease-free-survival [
11]. In TNBC cells, high levels of ERβ are associated with increased sensitivity to doxorubicin, which is controlled by the activation of the PI3K/AKT/mTOR pathway [
52]. Triggering MDA-MB-231 and BT549 cell lines, with a specific agonist (liquiritigenin) for ERβ, increases the sensitivity to doxorubicin, a chemotherapeutic agent. In both TNBC cell lines, combinatorial treatment with liquiritigenin and doxorubicin showed a stronger effect than that exerted by each drug in monotherapy. Analysis of the molecular mechanisms has shown that combinatorial treatment causes a strong inhibition of the PI3K/AKT/mTOR pathway and this mechanism is correlated to ERβ expression [
52].
Other studies have analyzed the role of ERβ in mediating the action of commonly used adjuvant drugs (such as tamoxifen or raloxifene) in TNBC cell lines. In MDA-MB-231 cells, ERβ expression fuels their overall effect or increases TNBC sensitivity to them. Since the mechanism of action of these drugs involves regulation of multiple signaling pathways, including EGFR, MAPK and PI3K, it has been reported that ERβ expression influences the effect of the aforementioned drugs, likely acting through a non-genomic way [
53]. Again, by acting through a non-genomic action, ERβ decreases cell survival in TNBC. In MDA-MB231 cells, ERβ stimulation or upregulation reduces cell survival and enhances apoptosis by activating the stress-regulated cell-death pathway at endoplasmic reticulum [
54]. Notably, by activating rapid actions, ERβ is also able to control the epithelial to mesenchymal transition (EMT) in TNBC. By overexpressing or downregulating ERβ in basal-like breast cancer cells (MDA-MB231 and Hs857T cells), the stabilization of an epithelial phenotype occurs, followed by inhibition of cell motility and invasion. These findings have been attributed to the ERβ-mediated upregulation of miR-200a/b/429, as well as the transcriptional repression of ZEB1 and SIP1, accounting for the increase in E-cadherin expression and inhibition of EMT. The direct correlation between ERβ and E-cadherin expression has been further confirmed in BC specimens. In the same experimental setting, Thomas and colleagues [
55] also showed that by reinforcing the interaction between EGFR and the ubiquitin ligase Cbl, ERβ increases the EGFR degradation and lowers the EGFR-mediated downstream signaling. In this way, ERβ further impairs EMT in TNBC. These findings were confirmed by in vivo experiments in zebrafish [
55].
Taken together, the data so far discussed underscore the relevance of ERβ2, ERβ5 and ERβ1 in TNBC. These studies also imply that quantification of the amount or relative ratios of the different ERβ isoforms might have prognostic and therapeutic relevance in TNBC. These considerations would help us for a better stratification of patients, although the question of ERβ ligand efficacy in TNBC still remains pending and agonism of ERβ in patients with advanced TNBC has shown a limited efficacy in phase 2 study [
31].
Figure 2 summarizes the aforementioned genomic and non-genomic pathways controlled by ERβ in TNBC.
Figure 2. Schematic representation of genomic and non-genomic actions of ERβ in TNBC. ERβ controls genomic and non-genomic actions upon ligand binding. By up-regulating or down-regulating the transcription of different genes, the receptor controls cell cycle, migration and invasion. Cytoplasmic ERβ rapidly activates the stress-regulated cell death pathway, or inhibits the PI3K/Akt/mTor pathway, or stimulates the EGFR degradation, thus promoting apoptosis, increasing the doxorubicin sensitivity and inhibiting the EMT transition and cell invasion, respectively.
5. Concluding Remarks
The pro-oncogenic properties of ERβ 2 and ERβ 5 suggest the possible development and clinical use of specific antagonists in treating TNBC. Lastly, the tight relationship between AR and ERβ in TNBC and the ability of ERβ to potentiate the antiandrogen effect is another aspect to be considered when developing therapeutic strategies in TNBC. Overall, the results collected and described in this review underscore the importance to ameliorate the detection methods for revealing ERβ. Only in this way, might we select the best and more tailored treatment for TNBC patients.
This entry is adapted from the peer-reviewed paper 10.3390/endocrines2030033