Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Mitochondria are the only animal cell organelles, except for the nucleus, with their own genetic information, called mitochondrial DNA (mtDNA). The mtDNA is a double-stranded, circular, 16,569 bp DNA molecule in humans, which codes 13 essential subunits of the respiratory chain complexes, 22 tRNAs, and two rRNAs, constituting part of the mitochondrial translation machinery.
- oocyte maturation
- mtDNA copy number
- mitochondria distribution
1. Introduction
In vitro maturation (IVM) technology is one of the most promising artificial reproductive technologies, as it allows fertility specialists to obtain oocytes without any or minimal hormonal stimulation on any day of the menstrual cycle [1]. It is mostly suitable for patients with polycystic ovarian morphology, oncological patients, or women with resistant ovary syndrome. This technology has finally surpassed its experimental status [1]; however, the clinical outcomes of the standard IVM programs are still inferior to the control ovarian stimulation programs [2]. It is believed that IVM oocytes have a decreased developmental potential due to cytoplasmic immaturity [3]. As soon as a cumulus–oocyte complex is released from a follicle, meiosis resumes. Yet, at the same time, the oocyte loses gap junctions with the cumulus cells and, therefore, supplementation with nutrients and signaling molecules stops, which leaves the cytoplasm immature. This means that the oocyte’s organelles have a structure and localization typical for the germinal vesicle (GV) stage of development.
One of the main cytoplasmic organelles are mitochondria, which are the source of energy production for the cells and control intracellular Ca2+ homeostasis [4][5]. In addition to their importance in energy production and calcium homeostasis, mitochondria also play a central role in many other essential functions of cells, including the regulation of cell death and signaling pathways, iron metabolism, and biosynthesis of some organic compounds [6][7][8]. During folliculogenesis, follicles undergo major growth, expanding approximately 500 times in size. The oocyte within the follicle also undergoes major structural and biochemical transformations and has to complete two meiotic divisions. Naturally, along with this energy-demanding process, the quantity and quality of mitochondria as well as their distribution pattern in an oocyte change [9].
2. Mitochondrial DNA Biology in Brief
Mitochondria are the only animal cell organelles, except for the nucleus, with their own genetic information, called mitochondrial DNA (mtDNA). The mtDNA is a double-stranded, circular, 16,569 bp DNA molecule in humans, which codes 13 essential subunits of the respiratory chain complexes, 22 tRNAs, and two rRNAs, constituting part of the mitochondrial translation machinery [10]. The mtDNA is stored in cells in nucleoids containing one copy of the genome linked with different nuclear-encoded proteins [11]. The nucleoid seems to be a layered structure in which replication and transcription occur in the central core, whereas translation and complex assembly occur in the peripheral region [12]. The mtDNA has some unique features in comparison to nuclear DNA: (i) multicopy nature of mtDNA with a broad range from several copies in sperm cells [13] to several thousand copies in the mature oocytes [14]; (ii) the mutagenesis rate of the mitochondrial genome is 20 times higher than the mutation rate of the nuclear DNA, with some unique features in its distribution across the genome [15]; and (iii) the multicopy nature of mtDNA allows the existence of mixed populations of mtDNA molecules—a phenomenon called heteroplasmy, when there is more than one mtDNA variant in a cell or an organism [16]. The clinical consequences of mutated mtDNA manifest only once a threshold of the mutant load is exceeded and these functional threshold levels vary, depending on the type of mtDNA mutation [17]. Any mtDNA mutation is supposed to be clinically manifested at the level of 70% heteroplasmy. However, it should be noted that this level is very conditional, and the relationship between the symptoms and the heteroplasmy level is very individual [18][19]. The heteroplasmy level can fluctuate from generation to generation. Hence, women harboring heteroplasmic mtDNA mutations can transmit a wide range of heteroplasmy levels to different offspring within the same sibship. The phenomenon was first shown by Hauswirth and Laipis on a Holstein cow and her offspring when the mtDNA allele variants rapidly shifted and became fixed in a few generations [20]. This is called a “mitochondrial bottleneck”, and it occurs owing to a profound dilution of mtDNA followed potentially by a selective replication of mtDNA genomes and asymmetric segregation of mitochondria. There are two hypotheses describing its time interval: (i) bottleneck as a result of the replication of a separate group of mtDNA during postpubertal folliculogenesis; and (ii) bottleneck during embryogenesis, when there is a significant decrease in the number of mtDNA copies [21][22][23][24].
3. Mitochondrial Dynamics in Oogenesis
Structural and morphological studies describe mitochondria from the primordial follicle stage to mature oocytes as naive, roundish-oval-like structures. Unlike mitochondria in differentiated somatic cells, which are mature and form highly structured networks, mitochondria in oocytes have unstructured cristae with a limited capacity for energy production [25]. Measurements of the total number of discrete mitochondria and of mtDNA in mouse oocytes indicate that, on average, each mitochondrion may carry only a single mtDNA copy [26]. To the best of our knowledge, there is no information on the relationship between the mitochondria count and mtDNA copy number in the mature human oocyte, which means that the study reported by Piko and Matsumoto [20] is the only one; everyone takes for granted the extrapolation from a mouse model to human oocytes. If the hypothesis is correct, then the defect mtDNA equals the defective mitochondrion, and an oocyte permits the quality control at the single-genome level by means of selective mitophagy. On the contrary, somatic cell mitochondria are assembled in networks (mitochondrions), and at each moment in time, an individual mitochondrion contains several nucleoids. This “dilution” of pathogenic mtDNA, together with functional mitochondrial complementation, makes the process of recognizing defective mitochondria less efficient in somatic cells [27].
During oocyte maturation, mtDNA copy number dramatically increases. Most likely, there are two turning points in the mtDNA copy number timeline on the segment from the primordial follicle to the MII oocyte. The first one is during the transition from the primordial to the primary follicle stage, and the second one is during the transition from GV to MII oocytes at the antral follicle stage. Primordial germ cells contain approximately 200 copies of mtDNA, while a mature oocyte possesses about 400,000 copies [28][29] (Figure 1). As noted earlier, such a significant increase in the mtDNA copies during postpubertal folliculogenesis may be one of the bottlenecks of mtDNA inheritance.
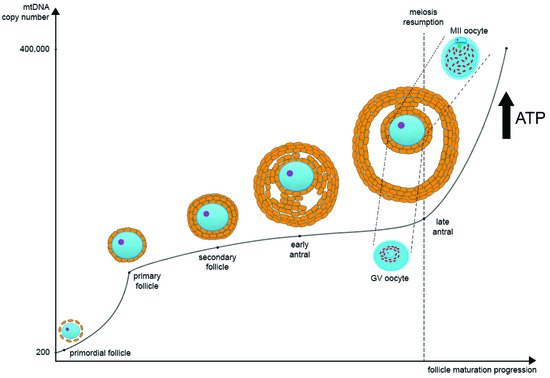
Figure 1. The regulation of mitochondrial DNA (mtDNA) copy number and mitochondria distribution during oocyte maturation. In primordial germ cells, the amount of mtDNA is very low. As oogenesis progresses, mtDNA copies increase significantly and then reach their maximum at the MII stage. There are two main points at which mtDNA copies go up very quickly: the first one is during the transition from the primordial to the primary follicle stage, and the second one is during the transition from GV to MII oocytes at the late antral follicle stage. In parallel as oogenesis progresses, ATP level also increases significantly as oocyte maturation requires a large amount of ATP for the subsequent transcription and translation. During the transition from GV to MII oocytes at the late antral follicle stage, the distribution of mitochondria changes significantly. They migrate from the center of the ooplasm to the pericortical region, distributing evenly through the whole ooplasm.
In recent years, we have begun to recognize the link between mtDNA replication and mitochondrial fission, which had previously been studied separately [30]. When mtDNA replication terminates, the molecules have to disconnect and separate into different mitochondria. It is known that nucleoids are fixed on the inner mitochondrial membrane (IMM), which helps in the separation of the last ones into different mitochondria during organelle division [31]. The exact protein composition of IMM and the nucleoid interaction site, as well as the detailed mechanism of the interconnection, are still to be recognized. However, it was shown that the loss of the mitochondrial contact site (MICOS) protein Mic60 results in disorganization of both the mitochondrial cristae and nucleotides [32]. It is possible that MICOS may have a role in anchoring mtDNA on the IMM surface in order to prevent their free diffusion around the separating mitochondria. Moreover, both ATAD3 and OPA1 were also colocalized with nucleoid components and have been proposed to be involved in the attachment of mtDNA to the IMM [33][34]. It is an open question whether the association of mtDNA with the IMM is stable or whether mtDNA attaches and separates from the membrane during DNA replication.
It still remains elusive as to which event, nucleoid replication or mitochondria fission, is primary. Mitochondrial morphology is coordinated with the cell cycle and promotes equal segregation of mitochondria during somatic cell division [35]. At the G1 stage of the somatic cell cycle, mitochondria have different morphologies. During the transition between the G1 and S, mitochondria start fusion, likely to enhance the production of ATP required for the S stage and the replication of nuclear DNA in particular. At the G2 and M stages, mitochondria start fission and look like individual organelles distributed throughout the cell [36]. In that condition, mitochondria are distributed between daughter cells. As oogonia cells divide by mitosis, the processes of mtDNA replication and organelle division are most likely similar to somatic cells. After oogonia become primary follicles, the mitotic division stops and the oocytes arrest at the diplotene stage of prophase I. Later on, in the segment between the primordial and primary follicle stage, an increase in both mtDNA copy number and mitochondria is observed [37]. During prophase I, the pairs of homologous chromosomes come together to form a bivalent, which contains four chromatids. Recombination can occur between any two chromatids within this structure which requires a huge amount of ATP [38]. There is another significant increase in the copy number of mtDNA prior to final oocyte maturation [37][39]. Most likely, such accumulation of mitochondria/mtDNA is necessary for further fertilization and further embryo development.
It is interesting that mtDNA replication events concur with the alterations in DNA methylation throughout oocyte maturation [40][41]. The correlation between the decreased DNA methylation levels at exon 2 in human POLG (DNA polymerase γ) and an increased mtDNA copy number for the glioblastoma multiforme cell line HSR-GBM1 and hNSCs derived from the NIH-approved human ESC line H9 was demonstrated [42]. We assume these findings could be extrapolated for oocyte maturation, as well as explain the mtDNA copy number changes. During oogenesis, DNA demethylation is mediated by the ten-eleven translocation methylcytosine dioxygenase (TET) family of enzymes. Interestingly, using the Western immunoblotting of samples from primary neuronal cultures, it was possible to find TET in the mitochondrial fraction [43]. It was also shown that demethylation of mtDNA results in an increase in copy number but not the number of mitochondrial transcripts, at least in glioblastoma cells [44]. Demethylation of nuclear and mtDNA likely occurs synchronously, but with different outcomes. DNA demethylation of nuclear-encoded mtDNA replication genes mediates their increased expression, whereas demethylation of the mitochondrial genome provides more templates for mtDNA replication. No changes in the number of mitochondrial transcripts would mean that no additional respiratory chain complexes anchor on the cristae, as mtDNA encodes the core parts of the oxidative phosphorylation complexes. This in turn contradicts the speculation that mitochondria in the oocyte play a key role in supplying the oocyte with ATP and indicates that mitochondria are only a means of mtDNA transmission to the next generation.
As nuclear maturation occurs in the oocyte, the distribution of mitochondria changes significantly. This distribution is very structured rather than chaotic [25][45][46]. According to the data using real-time confocal imaging, a transmission electron microscope, and ultrastructural analysis in in vitro matured oocytes, starting at the GV stage of an antral follicle and until 5 h before the germinal vesicle breakdown (GVB), mitochondria preferentially accumulate close to the perinuclear region and occupy about 80% of the cytoplasm. Then, after GVB and up to the MII stage, mitochondria are distributed equally in the ooplasm and occupy up to 90% of its volume [25][46]. It was shown in a mouse model that during meiosis I mitochondria localize near the spindle and then migrate with pairs of homologous chromosomes towards the pericortical ooplasm. As cytokinesis during meiosis is unequal, most mitochondria are moved back to the spindle poles and are excluded from the polar body 1 (PB1). The same mechanism happens with the extrusion of the second polar body (PB2) [47].
Different subpopulations of mitochondria can be distinguished by considering membrane potential within the oocyte. Highly polarized mitochondria exclusively occupy the pericortical region of the ooplasm, likely of importance to subsequent sperm penetration. Moreover, mitochondria can also differ in size and even nucleoid context within an oocyte [48][49]. Yet, the functional differences in mitochondrial subpopulations still remain to be determined. Furthermore, we believe that results obtained with fluorescent mitochondrial membrane potential probes should be dealt with using great caution. This kind of experimental data requires multiple controls and appropriate interpretation as non-protonic charges may have an effect on the dye behavior [50].
4. The Influence of Mitochondrial Function on Oocyte Quality
Female mammals at birth carry millions of primordial follicles containing oocytes arrested at the diplotene stage of prophase I, which constitute the follicular reserve of a female. As oocyte maturation requires a large amount of ATP for continuous transcription and translation, the availability of a sufficient number of functional mitochondria is crucial. However, since the mitochondria of immature oocytes are naive, it is likely that the energy to support oocyte maturation is provided mainly by the surrounding cumulus and granulosa cells [51]. Ovulating oocytes lose their connections with the cumulus cells that have provided them so far with energy and have to activate their own mitochondria. Thus, by the time of final oocyte maturation, a sufficient number of mitochondria have been accumulated. Deviation in mtDNA is associated with ovarian reserve reduction. A distinct range of oocyte mtDNA copy numbers were shown in patients with diminished ovarian reserve (100,000 ± 99,000 copies) compared to women with the normal ovarian reserve (318,000 ± 184,000 copies) [52]. The same pattern was also shown for the polar body mtDNA copy number [53]. These data indicate the importance of the number of mitochondria and mtDNA for oocyte quality.
Meiosis occurs twice during oocyte maturation, once during ovulation (meiosis I), and once during fertilization (meiosis II). Spindle microtubules are assembled from the opposite spindle poles and this process is extremely energy-intensive for oocytes in terms of ATP. There is evidence that decreases in ATP level result in meiotic errors that could alter the number of chromosomes in cells and thus lead to genetic disorders [38][47]. Such errors result in non-extrusion of the PB1, irregular distribution of chromosomes, and aneuploidy. Using HeLa and MDAMB 435 lines as a model system, it was shown that mitochondrial function is fundamental for maintaining the integrity of the genome by preventing chromosomal translocations and rearrangements [54]. Moreover, the correct spindle formation and the occurrence of chaotic mosaicism in human preimplantation embryos are in direct correlation with the value of the mitochondrial membrane potential [55]. ATP levels increase during the polar body extrusion and higher amounts of ATP correlate with a higher fertilization rate of mature oocytes [56].
Mitochondria not only regulate the energy metabolism but also the maintenance of the intracellular Ca2+ ion homeostasis [57]. The increasing Ca2+ concentration can disrupt oxidative phosphorylation and redox homeostasis or can result in the opening of the mitochondrial permeability transition pore which impairs the mitochondrial function and can cause apoptosis [58]. The low transfer of Ca2+ from the endoplasmic reticulum (ER) to the mitochondria contributes to the bioenergy crisis. In turn, disruption of mitochondrial oxidative phosphorylation (mPTP) and ATP production can affect the ATP-dependent calcium fluctuations. The entry of Ca2+ into mitochondria is stimulated by the activity of the respiratory chain complexes and causes the movement of protons into the intermembrane space, increasing the potential of the mitochondrial membrane [59].
mtDNA mutations can also result in disease and aging [60]. The study of point mutations and deletion of mtDNA in the oocytes of infertile or aged women has also been carried out. It has been shown that the number of de novo mtDNA mutations in children increases with maternal age [61][62], which might be attributed to oocyte aging. We have carefully estimated the mitochondrial mutation spectrum in offspring and mothers with a reconstruction of the de novo mitochondrial mutations that occurred in oocytes [15], and found that all the analyses of de novo mutations in mother–offspring pairs showed a trend of A > G rate increasing with oocyte’s time of dormancy. Thus, the primary oocyte arrested at the diplotene stage of prophase I can undergo accumulation of mtDNA rearrangements and accumulation of mutations during the long period of dormancy, even for several decades. Such mutations are asymmetric and likely are caused by the oxidation of mtDNA while the oocyte is waiting for fertilization [15]. It has been suggested that primary oocytes carrying deleterious mtDNA mutations are eliminated by follicular atresia, which may explain some cases of diminished ovarian reserve [63].
The purifying selection of pathogenic mtDNA during oocyte maturation is vitally important for offspring, as it can eliminate severe mtDNA mutations [64]. However, the current evidence for germline selection at the meiotic level comes from the incomplete sequencing of mtDNA in the first polar body. Interestingly, new data obtained from PB1 mtDNA deep sequencing revealed a high number of pathogenic mtDNA variants in comparison with its oocyte complement [65]. In order to further explore the transmission of defective mitochondria through generations, Sha et al. transferred the defective mitochondria in mouse germline, including the cumulus–oocyte complexes at the germinal vesicle and MII oocyte stages. The experiments confirmed that during the first and second meiosis, defective mitochondria are moved into PB1 and PB2. It was concluded that the two meiotic processes function as a kind of ultimate tier protection mechanism for future generations [65].
Thus, mitochondrial activity is a balance between the absolute mtDNA copy number, the number of mitochondria in a cell, functional capacity (mutational load), and organelle mobility.
This entry is adapted from the peer-reviewed paper 10.3390/cells10092484
References
- Practice Committees of the American Society for Reproductive Medicine, the Society of Reproductive Biologists and Technologists, and the Society for Assisted Reproductive Technology. In Vitro Maturation: A Committee Opinion. Fertil. Steril. 2021, 115, 298–304.
- Walls, M.L.; Hunter, T.; Ryan, J.P.; Keelan, J.A.; Nathan, E.; Hart, R.J. In Vitro Maturation as an Alternative to Standard in Vitro Fertilization for Patients Diagnosed with Polycystic Ovaries: A Comparative Analysis of Fresh, Frozen and Cumulative Cycle Outcomes. Hum. Reprod. 2015, 30, 88–96.
- De Vos, M.; Grynberg, M.; Ho, T.M.; Yuan, Y.; Albertini, D.F.; Gilchrist, R.B. Perspectives on the Development and Future of Oocyte IVM in Clinical Practice. J. Assist. Reprod. Genet. 2021, 38, 1265–1280.
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, Mitochondria and Cell Metabolism: A Functional Triangle in Bioenergetics. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2019, 1866, 1068–1078.
- Boyman, L.; Karbowski, M.; Lederer, W.J. Regulation of Mitochondrial ATP Production: Ca2+ Signaling and Quality Control. Trends Mol. Med. 2020, 26, 21–39.
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754.
- Lill, R.; Freibert, S.-A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499.
- Bock, F.J.; Tait, S.W.G. Mitochondria as Multifaceted Regulators of Cell Death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100.
- Van Blerkom, J. Mitochondrial Function in the Human Oocyte and Embryo and Their Role in Developmental Competence. Mitochondrion 2011, 11, 797–813.
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465.
- Kukat, C.; Wurm, C.A.; Spahr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-Resolution Microscopy Reveals That Mammalian Mitochondrial Nucleoids Have a Uniform Size and Frequently Contain a Single Copy of MtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539.
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2008, 283, 3665–3675.
- Song, G.J.; Lewis, V. Mitochondrial DNA Integrity and Copy Number in Sperm from Infertile Men. Fertil. Steril. 2008, 90, 2238–2244.
- Wai, T.; Ao, A.; Zhang, X.; Cyr, D.; Dufort, D.; Shoubridge, E.A. The Role of Mitochondrial DNA Copy Number in Mammalian Fertility1. Biol. Reprod. 2010, 83, 52–62.
- Mikhaylova, A.G.; Mikhailova, A.A.; Ushakova, K.; Tretiakov, E.O.; Shamansky, V.; Yurchenko, A.; Zazhytska, M.; Zdobnov, E.; Makeev, V.; Yurov, V.; et al. Mammalian Mitochondrial Mutational Spectrum as a Hallmark of Cellular and Organismal Aging. bioRxiv 2021.
- Stewart, J.B.; Chinnery, P.F. Extreme Heterogeneity of Human Mitochondrial DNA from Organelles to Populations. Nat. Rev. Genet. 2021, 22, 106–118.
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial Threshold Effects. Biochem. J. 2003, 370, 751–762.
- Lightowlers, R.; Chinnegy, P.F.; Turnbull, M.; Howell, N. Mammalian mitochondrial Genetics: Heredity, Heteroplasmy and Disease. Trends Genet. 1997, 13, 450–455.
- White, S.L.; Collins, V.R.; Wolfe, R.; Cleary, M.A.; Shanske, S.; DiMauro, S.; Dahl, H.-H.M.; Thorburn, D.R. Genetic Counseling and Prenatal Diagnosis for the Mitochondrial DNA Mutations at Nucleotide 8993. Am. J. Hum. Genet. 1999, 65, 474–482.
- Hauswirth, W.W.; Laipis, P.J. Mitochondrial DNA Polymorphism in a Maternal Lineage of Holstein Cows. Proc. Natl. Acad. Sci. USA 1982, 79, 4686–4690.
- Cao, L.; Shitara, H.; Horii, T.; Nagao, Y.; Imai, H.; Abe, K.; Hara, T.; Hayashi, J.-I.; Yonekawa, H. The Mitochondrial Bottleneck Occurs without Reduction of MtDNA Content in Female Mouse Germ Cells. Nat. Genet. 2007, 39, 386–390.
- Cree, L.M.; Samuels, D.C.; de Sousa Lopes, S.C.; Rajasimha, H.K.; Wonnapinij, P.; Mann, J.R.; Dahl, H.-H.M.; Chinnery, P.F. A Reduction of Mitochondrial DNA Molecules during Embryogenesis Explains the Rapid Segregation of Genotypes. Nat. Genet. 2008, 40, 249–254.
- Floros, V.I.; Pyle, A.; Dietmann, S.; Wei, W.; Tang, W.C.W.; Irie, N.; Payne, B.; Capalbo, A.; Noli, L.; Coxhead, J.; et al. Segregation of Mitochondrial DNA Heteroplasmy through a Developmental Genetic Bottleneck in Human Embryos. Nat. Cell Biol. 2018, 20, 144–151.
- Wai, T.; Teoli, D.; Shoubridge, E.A. The Mitochondrial DNA Genetic Bottleneck Results from Replication of a Subpopulation of Genomes. Nat. Genet. 2008, 40, 1484–1488.
- Trebichalská, Z.; Kyjovská, D.; Kloudová, S.; Otevřel, P.; Hampl, A.; Holubcová, Z. Cytoplasmic Maturation in Human Oocytes: An Ultrastructural Study. Biol. Reprod. 2021, 104, 106–116.
- Pikó, L.; Matsumoto, L. Number of Mitochondria and Some Properties of Mitochondrial DNA in the Mouse Egg. Dev. Biol. 1976, 49, 1–10.
- Hoitzing, H.; Johnston, I.G.; Jones, N.S. What Is the Function of Mitochondrial Networks? A Theoretical Assessment of Hypotheses and Proposal for Future Research. BioEssays 2015, 37, 687–700.
- Jansen, R.P.S. Germline Passage of Mitochondria: Quantitative Considerations and Possible Embryological Sequelae. Hum. Reprod. 2000, 15, 112–128.
- Santos, T.A.; El Shourbagy, S.; St. John, J.C. Mitochondrial Content Reflects Oocyte Variability and Fertilization Outcome. Fertil. Steril. 2006, 85, 584–591.
- Nicholls, T.J.; Gustafsson, C.M. Separating and Segregating the Human Mitochondrial Genome. Trends Biochem. Sci. 2018, 43, 869–881.
- Ban-Ishihara, R.; Ishihara, T.; Sasaki, N.; Mihara, K.; Ishihara, N. Dynamics of Nucleoid Structure Regulated by Mitochondrial Fission Contributes to Cristae Reformation and Release of Cytochrome c. Proc. Natl. Acad. Sci. USA 2013, 110, 11863–11868.
- Li, H.; Ruan, Y.; Zhang, K.; Jian, F.; Hu, C.; Miao, L.; Gong, L.; Sun, L.; Zhang, X.; Chen, S.; et al. Mic60/Mitofilin Determines MICOS Assembly Essential for Mitochondrial Dynamics and MtDNA Nucleoid Organization. Cell Death Differ. 2016, 23, 380–392.
- Elachouri, G.; Vidoni, S.; Zanna, C.; Pattyn, A.; Boukhaddaoui, H.; Gaget, K.; Yu-Wai-Man, P.; Gasparre, G.; Sarzi, E.; Delettre, C.; et al. OPA1 Links Human Mitochondrial Genome Maintenance to MtDNA Replication and Distribution. Genome Res. 2011, 21, 12–20.
- He, J.; Mao, C.-C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ Protein ATAD3 Has Displacement Loop Binding Properties and Is Involved in Mitochondrial Nucleoid Organization. J. Cell Biol. 2007, 176, 141–146.
- Mishra, P.; Chan, D.C. Mitochondrial Dynamics and Inheritance during Cell Division, Development and Disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646.
- Martínez-Diez, M.; Santamaría, G.; Ortega, Á.D.; Cuezva, J.M. Biogenesis and Dynamics of Mitochondria during the Cell Cycle: Significance of 3′UTRs. PLoS ONE 2006, 1, e107.
- Cotterill, M.; Harris, S.E.; Collado Fernandez, E.; Lu, J.; Huntriss, J.D.; Campbell, B.K.; Picton, H.M. The Activity and Copy Number of Mitochondrial DNA in Ovine Oocytes throughout Oogenesis in Vivo and during Oocyte Maturation in Vitro. MHR: Basic Sci. Reprod. Med. 2013, 19, 444–450.
- Zhang, X.; Wu, X.Q.; Lu, S.; Guo, Y.L.; Ma, X. Deficit of Mitochondria-Derived ATP during Oxidative Stress Impairs Mouse MII Oocyte Spindles. Cell Res. 2006, 16, 841–850.
- Mao, J.; Whitworth, K.M.; Spate, L.D.; Walters, E.M.; Zhao, J.; Prather, R.S. Regulation of Oocyte Mitochondrial DNA Copy Number by Follicular Fluid, EGF, and Neuregulin 1 during in Vitro Maturation Affects Embryo Development in Pigs. Theriogenology 2012, 78, 887–897.
- St. John, J.C. Mitochondria and Female Germline Stem Cells—A Mitochondrial DNA Perspective. Cells 2019, 8, 852.
- St. John, J.C. Epigenetic Regulation of the Nuclear and Mitochondrial Genomes: Involvement in Metabolism, Development, and Disease. Annu. Rev. Anim. Biosci. 2021, 9, 203–224.
- Lee, W.; Johnson, J.; Gough, D.J.; Donoghue, J.; Cagnone, G.L.M.; Vaghjiani, V.; Brown, K.A.; Johns, T.G.; St. John, J.C. Mitochondrial DNA Copy Number Is Regulated by DNA Methylation and Demethylation of POLGA in Stem and Cancer Cells and Their Differentiated Progeny. Cell Death Dis. 2015, 6, e1664.
- Dzitoyeva, S.; Chen, H.; Manev, H. Effect of Aging on 5-Hydroxymethylcytosine in Brain Mitochondria. Neurobiol. Aging 2012, 33, 2881–2891.
- Sun, X.; Johnson, J.; St. John, J.C. Global DNA Methylation Synergistically Regulates the Nuclear and Mitochondrial Genomes in Glioblastoma Cells. Nucleic Acids Res. 2018, 46, 5977–5995.
- Liu, S.; Li, Y.; Feng, H.L.; Yan, J.H.; Li, M.; Ma, S.Y.; Chen, Z.J. Dynamic Modulation of Cytoskeleton during in Vitro Maturation in Human Oocytes. Am. J. Obstet. Gynecol. 2010, 203, 151.e1–151.e7.
- Takahashi, Y.; Hashimoto, S.; Yamochi, T.; Goto, H.; Yamanaka, M.; Amo, A.; Matsumoto, H.; Inoue, M.; Ito, K.; Nakaoka, Y.; et al. Dynamic Changes in Mitochondrial Distribution in Human Oocytes during Meiotic Maturation. J. Assist. Reprod. Genet. 2016, 33, 929–938.
- Dalton, C.M.; Carroll, J. Biased Inheritance of Mitochondria during Asymmetric Cell Division in the Mouse Oocyte. J. Cell Sci. 2013, 126, 2955–2964.
- Woods, D.; Khrapko, K.; Tilly, J. Influence of Maternal Aging on Mitochondrial Heterogeneity, Inheritance, and Function in Oocytes and Preimplantation Embryos. Genes 2018, 9, 265.
- Van Blerkom, J. Mitochondria in Human Oogenesis and Preimplantation Embryogenesis: Engines of Metabolism, Ionic Regulation and Developmental Competence. Reproduction 2004, 128, 269–280.
- Perry, S.W.; Norman, J.P.; Barbieri, J.; Brown, E.B.; Gelbard, H.A. Mitochondrial Membrane Potential Probes and the Proton Gradient: A Practical Usage Guide. BioTechniques 2011, 50, 98–115.
- Dumollard, R.; Campbell, K.; Halet, G.; Carroll, J.; Swann, K. Regulation of Cytosolic and Mitochondrial ATP Levels in Mouse Eggs and Zygotes. Dev. Biol. 2008, 316, 431–440.
- May-Panloup, P.; Chrétien, M.F.; Jacques, C.; Vasseur, C.; Malthièry, Y.; Reynier, P. Low Oocyte Mitochondrial DNA Content in Ovarian Insufficiency. Hum. Reprod. 2005, 20, 593–597.
- Konstantinidis, M.; Alfarawati, S.; Hurd, D.; Paolucci, M.; Shovelton, J.; Fragouli, E.; Wells, D. Simultaneous Assessment of Aneuploidy, Polymorphisms, and Mitochondrial DNA Content in Human Polar Bodies and Embryos with the Use of a Novel Microarray Platform. Fertil. Steril. 2014, 102, 1385–1392.
- Desler, C.; Munch-Petersen, B.; Stevnsner, T.; Matsui, S.-I.; Kulawiec, M.; Singh, K.K.; Rasmussen, L.J. Mitochondria as Determinant of Nucleotide Pools and Chromosomal Stability. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2007, 625, 112–124.
- Wilding, M.; De Placido, G.; De Matteo, L.; Marino, M.; Alviggi, C.; Dale, B. Chaotic Mosaicism in Human Preimplantation Embryos Is Correlated with a Low Mitochondrial Membrane Potential. Fertil. Steril. 2003, 79, 340–346.
- Zhao, J.; Li, Y. Adenosine Triphosphate Content in Human Unfertilized Oocytes, Undivided Zygotes and Embryos Unsuitable for Transfer or Cryopreservation. J. Int. Med. Res. 2012, 40, 734–739.
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283.
- Baumgartner, H.K.; Gerasimenko, J.V.; Thorne, C.; Ferdek, P.; Pozzan, T.; Tepikin, A.V.; Petersen, O.H.; Sutton, R.; Watson, A.J.M.; Gerasimenko, O.V. Calcium Elevation in Mitochondria Is the Main Ca2+ Requirement for Mitochondrial Permeability Transition Pore (MPTP) Opening. J. Biol. Chem. 2009, 284, 20796–20803.
- Fink, B.D.; Bai, F.; Yu, L.; Sivitz, W.I. Regulation of ATP Production: Dependence on Calcium Concentration and Respiratory State. Am. J. Physiol.—Cell Physiol. 2017, 313, C146–C153.
- Wallace, D.C. Mitochondrial DNA Mutations in Disease and Aging. Environ. Mol. Mutagen. 2010, 51, 440–450.
- Rebolledo-Jaramillo, B.; Su, M.S.-W.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal Age Effect and Severe Germ-Line Bottleneck in the Inheritance of Human Mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479.
- Arbeithuber, B.; Hester, J.; Cremona, M.A.; Stoler, N.; Zaidi, A.; Higgins, B.; Anthony, K.; Chiaromonte, F.; Diaz, F.J.; Makova, K.D. Age-Related Accumulation of de Novo Mitochondrial Mutations in Mammalian Oocytes and Somatic Tissues. PLoS Biol. 2020, 18, e3000745.
- Colnaghi, M.; Pomiankowski, A.; Lane, N. The Need for High-Quality Oocyte Mitochondria at Extreme Ploidy Dictates Germline Development. Elife 2021, 10, e69344.
- Stewart, J.B.; Freyer, C.; Elson, J.L.; Larsson, N.-G. Purifying Selection of MtDNA and Its Implications for Understanding Evolution and Mitochondrial Disease. Nat. Rev. Genet. 2008, 9, 657–662.
- Sha, H.; Yang, Y.; Shi, S.; Ji, D.; Pan, J. Germline Selection by Meiosis Defends the Transmission of Defective Mitochondria with MtDNA Variants. bioRxiv 2020.
This entry is offline, you can click here to edit this entry!