Atherosclerosis is a multifactorial chronic disease that has a prominent inflammatory component. Currently, atherosclerosis is regarded as an active autoimmune process that involves both innate and adaptive immune pathways. One of the drivers of this process is the presence of modified low-density lipoprotein (LDL). For instance, lipoprotein oxidation leads to the formation of oxidation-specific epitopes (OSE) that can be recognized by the immune cells. Macrophage response to OSEs is recognized as a key trigger for initiation and a stimulator of progression of the inflammatory process in the arteries. At the same time, the role of oxidized LDL components is not limited to pro-inflammatory stimulation, but includes immunoregulatory effects that can have protective functions.
- atherosclerosis
- LDL
- oxidized LDL
- macrophage
- inflammation
- immunomodulation
1. Introduction
The multifactorial nature of atherosclerosis pathogenesis makes studying this disease challenging. According to current view, atherosclerosis can be considered as a chronic inflammatory disease associated with progressive accumulation of lipids and inflammatory cells in the arterial wall. The atherogenic process begins with deposition of low-density lipoprotein (LDL), which is normally present in the blood plasma, in the subendothelial space of the arterial wall. In human arteries, such deposition often occurs at the sites of laminar flow perturbation, where activated or dysfunctional endothelial cells are present [1].
For many years, LDL was known as the main source of lipid accumulation in atherosclerosis, and much of anti-atherosclerotic therapies is aimed at correcting the blood lipid profile to slow down the disease progression.
2. Pathways of Lipid Oxidation and Their Relevance for Atherosclerosis
The presence of activated endothelial vascular cells, neutrophils, macrophages and T and B cells in atherosclerotic plaques, together with the proinflammatory cytokine environment, suggests that atherosclerosis is an active immunopathological process [1]. The hypothesis of oxLDL acting as a trigger of atherosclerosis development originated from the studies in 1980s–1990s that showed that macrophages treated with oxLDL, but not native LDL, accumulated cholesterol esters [8]. Another study demonstrated the presence of autoantibody response to oxLDL in apolipoprotein E deficient mice [9]. Since then, it has become clear that atherosclerosis is an autoimmune process and oxidized forms of LDL are among most validated autoantigens relevant to atheroinflammation [1].
3. Receptor-Mediated Uptake and Effects of Oxidation-Specific Epitopes
3.1. Interaction with CD36
| PRR | OSE | Effect | Cells |
|---|---|---|---|
| Scavenger receptors | |||
| SR-A1,2 | MDA | Uptake | Macrophages, mast, dendritic, endothelial, smooth muscle cells |
| SR-B1 | PC-OxPL | Uptake | Monocytes/macrophages, hepatocytes, adipocytes |
| SRECI/II | OxLDL | Uptake | Endothelial cells, macrophages, CD8+ cells |
| SR-PSOX | Ox-PS | Uptake Foam cell formation |
Macrophages, smooth muscle, dendritic, and endothelial cells, and B-cells and T cells |
| LOX-1 | MDA | Monocyte adhesion Uptake Inflammation |
Endothelial and smooth muscle cells, macrophages, platelets |
| 4-HNE | |||
| CD36 | PC-OxPL | Uptake Inflammation |
Macrophages, platelets, adipocytes, epithelial and endothelial cells |
| OxPS | Uptake Inflammation |
||
| CEP | Uptake Inflammation |
||
| TLRs | |||
| TLRs 4-6 | PC-OxPL | Inflammation | Monocytes/macrophages, dendritic cells, mast cells, B cells |
| TLR4 | OxCE | Inflammation Foam cell formation |
Monocytes/macrophages, dendritic cells, mast cells, B cells |
| OxPE | Inflammation Foam cell formation |
||
| 4-HNE | Inflammation | ||
| TLRs 2-6 | CEP | Inflammation Thrombosis |
Monocytes/macrophages, dendritic cells, mast cells, B cells, platelets |
| OxPL | Angiogenesis ER stress |
||
| TLR9 | CEP | Promotion of platelet hyperreactivity and thrombosis | Platelets |
| Complement | |||
| CFH | MDA | Neutralization Opsonization |
|
| C3a | MDA | Complement activation | |
| CRP | PC-OxPL | Enhanced efferocytosis | |
| Other PRRs | |||
| MFG-E8 | OxPS | Enhanced efferocytosis | |
| OxPE | Enhanced efferocytosis | ||
| Annexin A5 | OxCL | Neutralization | |
| CD16 | MDA | Inflammation | Macrophages |
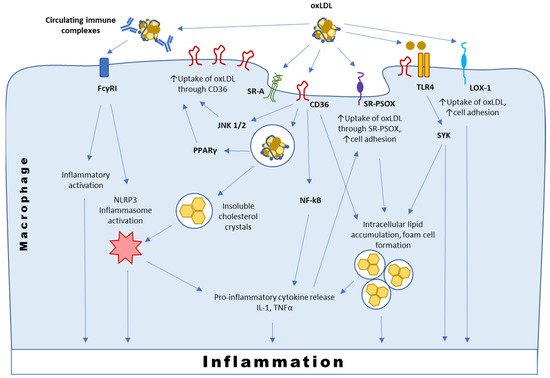
3.2. Interaction with SR-PSOX
3.3. Interaction with Immunoglobulins and TLRs
3.4. Interaction with LOX-1 and Other Scavenger Receptors
3.5. Interaction with Soluble Factors and Chaperons
OSE can also be sensed by soluble factors: Complement factor H (CFH), C3a, C-reactive protein (CRP), Annexin A5 and others, including innate natural antibodies [9]. These interactions may lead to complement activation as well as opsonization and enhanced efferocytosis of OSE-bearing targets. CFH is the major inhibitor of the alternative pathway of complement activation that was recently shown to bind to and prevent the proinflammatory effects of MDA epitopes [28]. CRP-oxLDL interaction triggers complement activation and enhances binding of oxLDL-antibody complexes to Fcγ receptors expressed on macrophages. CRP and modified LDL are colocalized in early atherosclerotic lesions of humans with coronary artery disease [5]. Expression of CRP is a characteristic feature of M1 pro-inflammatory macrophages [29]. In atherosclerotic plaques, CRP participates in a positive feedback loop with oxLDL, whereby increased levels of oxLDL induce endothelial cells and macrophages to express CRP, which may in turn increase the expression of LOX-1 to promote the uptake of atherogenic LDL into cells [30]. Involvement of complement in the pathogenesis of atherosclerosis is being actively studied and was recently discussed elsewhere [6][7].
4. Anti-Inflammatory and Immunoregulatory Functions of oxLDL
5. OxLDL as a Macrophage Polarization Signal
In the plaque environment, macrophages are exposed to various signals and stimuli, including cytokines, modified lipids, senescent erythrocytes and hypoxia, that influence their transcriptional program and functional phenotype [4]. As a consequence, intraplaque macrophages undergo polarization to distinct subtypes playing opposite roles in atherosclerosis pathology. The early classification of macrophages to M1 (proinflammatory) and M2 (anti-inflammatory) phenotypes is currently regarded as oversimplified and outdated, but can still be useful, especially for interpreting the results of in vitro studies [36].
6. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9080915
References
- Cinoku, I.I.; Mavragani, C.P.; Moutsopoulos, H.M. Atherosclerosis: Beyond the lipid storage hypothesis. The role of autoimmunity. Eur. J. Clin. Investig. 2020, 50, e13195.
- Gianazza, E.; Brioschi, M.; Martinez Fernandez, A.; Casalnuovo, F.; Altomare, A.; Aldini, G.; Banfi, C. Lipid Peroxidation in Atherosclerotic Cardiovascular Diseases. Antioxid. Redox Signal. 2021, 34, 49–98.
- Chinetti-Gbaguidi, G.; Colin, S.; Staels, B. Macrophage subsets in atherosclerosis. Nat. Rev. Cardiol. 2015, 12, 10–17.
- Orekhov, A.N.; Nikiforov, N.G.; Sukhorukov, V.N.; Kubekina, M.V.; Sobenin, I.A.; Wu, W.-K.; Foxx, K.K.; Pintus, S.; Stegmaier, P.; Stelmashenko, D.; et al. Role of Phagocytosis in the Pro-Inflammatory Response in LDL-Induced Foam Cell Formation; a Transcriptome Analysis. Int. J. Mol. Sci. 2020, 21, 817.
- Bhakdi, S.; Torzewski, M.; Klouche, M.; Hemmes, M. Complement and atherogenesis: Binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2348–2354.
- Martin-Ventura, J.L.; Martinez-Lopez, D.; Roldan-Montero, R.; Gomez-Guerrero, C.; Blanco-Colio, L.M. Role of complement system in pathological remodeling of the vascular wall. Mol. Immunol. 2019, 114, 207–215.
- Hovland, A.; Jonasson, L.; Garred, P.; Yndestad, A.; Aukrust, P.; Lappegård, K.T.; Espevik, T.; Mollnes, T.E. The complement system and toll-like receptors as integrated players in the pathophysiology of atherosclerosis. Atherosclerosis 2015, 241, 480–494.
- Quinn, M.T.; Parthasarathy, S.; Fong, L.G.; Steinberg, D. Oxidatively modified low density lipoproteins: A potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proc. Natl. Acad. Sci. USA 1987, 84, 2995–2998.
- Palinski, W.; Hörkkö, S.; Miller, E.; Steinbrecher, U.P.; Powell, H.C.; Curtiss, L.K.; Witztum, J.L. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J. Clin. Investig. 1996, 98, 800–814.
- Shibata, T.; Shimizu, K.; Hirano, K.; Nakashima, F.; Kikuchi, R.; Matsushita, T.; Uchida, K. Adductome-based identification of biomarkers for lipid peroxidation. J. Biol. Chem. 2017, 292, 8223–8235.
- Uchida, K.; Shibata, T.; Toyokuni, S.; Daniel, B.; Zarkovic, K.; Zarkovic, N.; Sasson, S. Development of a novel monoclonal antibody against 4-hydroxy-2E,6Z-dodecadienal (4-HDDE)-protein adducts: Immunochemical application in quantitative and qualitative analyses of lipid peroxidation in vitro and ex vivo. Free Radic. Biol. Med. 2018, 124, 12–20.
- Fredrikson, G.N.; Hedblad, B.; Berglund, G.; Alm, R.; Ares, M.; Cercek, B.; Chyu, K.Y.; Shah, P.K.; Nilsson, J. Identification of immune responses against aldehyde-modified peptide sequences in apoB associated with cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 872–878.
- Duner, P.; To, F.; Alm, R.; Gonçalves, I.; Fredrikson, G.N.; Hedblad, B.; Berglund, G.; Nilsson, J.; Bengtsson, E. Immune responses against fibronectin modified by lipoprotein oxidation and their association with cardiovascular disease. J. Intern. Med. 2009, 265, 593–603.
- Vallejo, J.; Dunér, P.; To, F.; Engelbertsen, D.; Gonçalves, I.; Nilsson, J.; Bengtsson, E. Activation of immune responses against the basement membrane component collagen type IV does not affect the development of atherosclerosis in ApoE-deficient mice. Sci. Rep. 2019, 9, 5964.
- Vallejo, J.; Dunér, P.; Fredrikson, G.N.; Nilsson, J.; Bengtsson, E.J. Autoantibodies against aldehyde-modified collagen type IV are associated with risk of development of myocardial infarction. Intern. Med. 2017, 282, 496–507.
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497.
- Park, Y.M. CD36, a scavenger receptor implicated in atherosclerosis. Exp. Mol. Med. 2014, 46, e99.
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008, 78, 185–196.
- Marleau, S.; Harb, D.; Bujold, K.; Avallone, R.; Iken, K.; Wang, Y.; Demers, A.; Sirois, M.G.; Febbraio, M.; Silverstein, R.L.; et al. EP 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein E-deficient mice from developing atherosclerotic lesions. FASEB J. 2005, 19, 1869–1871.
- Bujold, K.; Mellal, K.; Zoccal, K.F.; Rhainds, D.; Brissette, L.; Febbraio, M.; Marleau, S.; Ong, H. EP 80317, a CD36 selective ligand, promotes reverse cholesterol transport in apolipoprotein E-deficient mice. Atherosclerosis 2013, 229, 408–414.
- Hofnagel, O.; Engel, T.; Severs, N.J.; Robenek, H.; Buers, I. SR-PSOX at sites predisposed to atherosclerotic lesion formation mediates monocyte-endothelial cell adhesion. Atherosclerosis 2011, 217, 371–378.
- Minami, M.; Kume, N.; Shimaoka, T.; Kataoka, H.; Hayashida, K.; Akiyama, Y.; Nagata, I.; Ando, K.; Nobuyoshi, M.; Hanyuu, M.; et al. Expression of SR-PSOX, a novel cell-surface scavenger receptor for phosphatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1796–1800.
- Zhang, L.; Liu, H.J.; Li, T.J.; Yang, Y.; Guo, X.L.; Wu, M.C.; Rui, Y.C.; Wei, L.X. Lentiviral vector-mediated siRNA knockdown of SR-PSOX inhibits foam cell formation in vitro. Acta Pharmacol. Sin. 2008, 29, 847–852.
- Tertov, V.V.; Sobenin, I.A.; Orekhov, A.N.; Jaakkola, O.; Solakivi, T.; Nikkari, T. Characteristics of low density lipoprotein isolated from circulating immune complexes. Atherosclerosis 1996, 122, 191–199.
- Lopes-Virella, M.F.; McHenry, M.B.; Lipsitz, S.; Yim, E.; Wilson, P.F.; Lackland, D.T.; Lyons, T.; Jenkins, A.J.; Virella, G.; DCCT/EDIC Research Group. Immune complexes containing modified lipoproteins are related to the progression of internal carotid intima-media thickness in patients with type 1 diabetes. Atherosclerosis 2007, 190, 359–369.
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86.
- Chen, H.; Li, D.; Sawamura, T.; Inoue, K.; Mehta, J.L. Upregulation of LOX-1 expression in aorta of hypercholesterolemicrabbits: Modulation by losartan. Biochem. Biophys. Res. Commun. 2000, 276, 1100–1104.
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.; Charbel Issa, P.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81.
- Kaplan, M.; Shur, A.; Tendler, Y. M1 Macrophages but Not M2 Macrophages Are Characterized by Upregulation of CRP Expression via Activation of NFkappaB: A Possible Role for Ox-LDL in Macrophage Polarization. Inflammation 2018, 41, 1477–1487.
- Stancel, N.; Chen, C.C.; Ke, L.Y.; Chu, C.S.; Lu, J.; Sawamura, T.; Chen, C.H. Interplay between CRP, Atherogenic LDL, and LOX-1 and Its Potential Role in the Pathogenesis of Atherosclerosis. Clin. Chem. 2016, 62, 320–327.
- Feige, E.; Yacov, N.; Salem, Y.; Levi, I.; Mendel, I.; Propheta-Meiran, O.; Shoham, A.; Hait-Darshan, R.; Polonsky, O.; George, J.; et al. Inhibition of monocyte chemotaxis by VB-201, a small molecule lecinoxoid, hinders atherosclerosis development in ApoE−/− mice. Atherosclerosis 2013, 229, 430–439.
- Mendel, I.; Feige, E.; Yacov, N.; Salem, Y.; Levi, I.; Propheta-Meiran, O.; Shoham, A.; Ishai, E.; George, J.; Harats, D.; et al. VB-201, an oxidized phospholipid small molecule, inhibits CD14- and Toll-like receptor-2-dependent innate cell activation and constrains atherosclerosis. Clin. Exp. Immunol. 2014, 175, 126–137.
- Doran, A.C.; Yurdagul, A., Jr.; Tabas, I. Efferocytosis in health and disease. Nat. Rev. Immunol. 2020, 20, 254–267.
- Martinet, W.; Kockx, M.M. Apoptosis in atherosclerosis: Focus on oxidized lipids and inflammation. Curr. Opin. Lipidol. 2001, 12, 535–541.
- Schrijvers, D.M.; De Meyer, G.R.; Kockx, M.M.; Herman, A.G.; Martinet, W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1256–1261.
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964.
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361.
- Han, X.; Ma, W.; Zhu, Y.; Sun, X.; Liu, N. Advanced glycation end products enhance macrophage polarization to the M1 phenotype via the HIF-1alpha/PDK4 pathway. Mol. Cell. Endocrinol. 2020, 514, 110878.
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam cell formation: A new target for fighting atherosclerosis and cardiovascular disease. Vasc. Pharmacol. 2019, 112, 54–71.