1. Introduction to the Glucosinolates
Glucosinolates are amino acid-derived plant-specialized metabolites that are largely found within the members of the family Brassicaceae, which includes vegetables such as broccoli, cabbage, and mustard, as well as the model plant
Arabidopsis thaliana (thale cress) [
1]. They have been reported in 14 other families from the order Capparales, as well as in the family Euphorbiaceae from the genus
Drypetes, which is unrelated to other glucosinolate-containing families [
2]. Glucosinolates can be classified according to their precursor amino acids. The aliphatic glucosinolates are derived from methionine, alanine, leucine, isoleucine, or valine; aromatic glucosinolates are built from phenylalanine or tyrosine; and the indole glucosinolates originate with tryptophan. Each of class of glucosinolate shares a core chemical structure consisting of a β-D-glucosyl residue linked to a (Z)-N-hydroximinosulfate ester through a sulfur and a variable amino acid-derived R group (
Figure 1) [
1]. To date, more than 130 glucosinolate molecules, of which
Arabidopsis contains 40 mainly derived from methionine and tryptophan, have been described [
3].
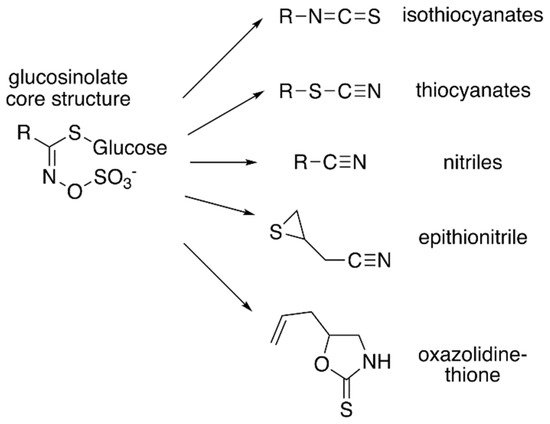
Figure 1. Core glucosinolate structure and hydrolysis product diversity. The core chemical structure of glucosinolates (left) consists of a β–D–glucosyl residue linked via a sulfur to a (Z)–N–hydroximinosulfate ester with and a variable amino acid-derived R group. Modifications of the aliphatic, aromatic, or indole R-group leads to the chemical diversity of this class of specialized metabolites. Hydrolysis of various glucosinolates leads to an array of bioactive molecules (right), including isothiocyanates, thiocyanates, nitriles, epithionitrile, and oxazolidine-thione.
Although the biological activities of all glucosinolates are yet to be elucidated, glucosinolates are believed to serve as responses to a wide range of external and/or environmental stimuli [
4,
5]. Much of glucosinolate function is related to their hydrolysis products (
Figure 1), which accumulate in response to plant tissue damage. The various hydrolysis products are generated by a thioglucoside glucohydrolase known as myrosinase, which hydrolyzes the glucose moiety of the core glucosinolate structure [
6,
7]. The resulting products are glucose and an unstable aglycone compound that can rearrange to form isothiocyanates, nitriles, or other hydrolysis products, based on the starting glucosinolate structure type (
Figure 1). The hydrolysis activity appears to be hindered by the physical separation of glucosinolates and myrosinase in intact plant tissue [
1]. Glucosinolate hydrolysis products are involved in communicating a range of information pertaining to plant defense against insects, bacteria, and fungi. Some hydrolysis products, such as isothiocyanates, can be hydrolyzed further by the phenylalanine ammonia lyase to generate toxic compounds that can be injurious to certain pathogens [
4]. Hence, some studies have proposed that glucosinolates may have a more direct role in plant defense.
Interest in glucosinolates has long been present in human society, mainly due to the distinct taste and flavors of certain Brassicaceae vegetables (cabbage, cauliflower, broccoli) and condiments (mustard, horseradish, wasabi) that are present in our diet [
8,
9]. Glucosinolates have gained significance in an agricultural sense with the increased importance of rapeseeds/canola (
Brassica napus,
B. rapa, and
B. juncea) as oil crops worldwide [
1]. To increase plant use efficiency, plant breeders have reduced the levels of glucosinolates in Brassica oil crops to allow the seedcake (i.e., the protein-rich residue left after seed processing) to be used as a nutritional supplement in animal feed [
10]. This is done to avoid the anti-nutritive effects that glucosinolates can have on animals, as one of the major glucosinolates in canola hydrolyzes to an oxazolidine-2-thione, which causes goiter and has other negative effects in cattle [
11]. Glucosinolate-rich plants can also be used as biofumigation agents. For example, post-harvest plant material can be incorporated into soils, where the glucosinolate-containing material can suppress pathogen, nematode, and/or weed growth [
12,
13,
14]. Breeders have also tried to modify glucosinolate levels in rapeseed foliage to address damage from fungal and insect pests [
15,
16]. For human health, glucosinolates are potentially useful because of their reported cancer-preventative action in animal models. For example, 4-methylsulfinylbutyl glucosinolate, which is found in broccoli, hydrolyzes to the isothiocyanate sulforaphane, a molecule that blocks the cell cycle and promotes apoptosis to fight tumor growth [
8,
17,
18,
19,
20]. Sulforaphane can also slow effects of
Helicobacter pylori-caused gastritis and stomach cancer [
21]. Overall, glucosinolate engineering in plants and production platforms such as bacteria offer useful tools for the application of these natural products in plant defense, agriculture, human health, and animal nutrition.
2. Glucosinolate Biosynthesis
Biosynthesis of glucosinolates requires the integration of multiple building blocks to generate their shared chemical structure. For the aliphatic glucosinolates, methionine provides the starting point of their biosynthesis through an iterative series of reactions that elongate the R-group, i.e., aliphatic chain elongation (
Figure 2). In the second stage of aliphatic glucosinolate synthesis (
Figure 3), the methionine-derived elongation products then undergo a series of modifications, including addition of the sugar and sulfate groups, to yield the core glucosinolate structure. A similar set of reactions for the aromatic and indole glucosinolates builds directly from the initial amino acids. Once the core glucosinolate structure is assembled, the third stage of biosynthesis consists of various secondary modifications to the R-group generate the diverse array of described glucosinolates [
22,
23,
24,
25,
26,
27].
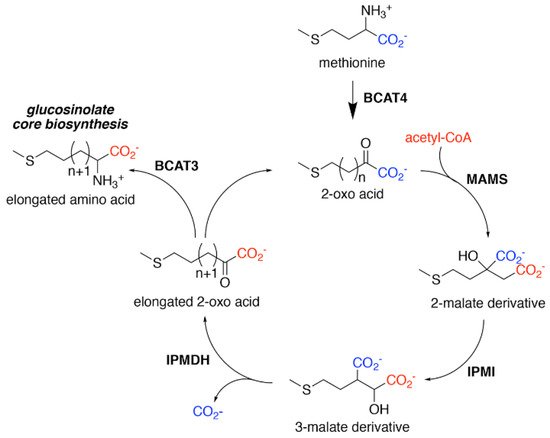
Figure 2. Aliphatic Glucosinolate Chain Elongation. Conversion of methionine to its corresponding 2-oxo acid (4–(methylsulfanyl)–2–oxobutanoate) by branched-chain amino transferase 4 (BCAT4) provides the starting point for the chain elongation cycle. A series of reactions catalyzed by methylthioalkylmalate synthase (MAMS), isopropylmalate isomerase (IPMI), and isopropylmalate dehydrogenase (IPMDH) result in elongation of the aliphatic chain by a single methylene group. The elongated 2–oxo can undergo transamination to the corresponding amino acid by BCAT3 or reenter the chain elongation cycle.
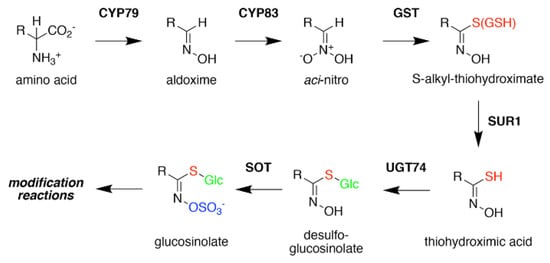
Figure 3. Glucosinolate Core Biosynthesis. A variety of amino acids (i.e., variable R-group), including elongated aliphatic methionine-derived molecules (see Figure 2), can be converted to aldoximes by members of the CYP79 cytochrome P450 family to begin construction of the core glucosinolate scaffold (see Figure 1, left). Members of the CYP83 family generate the unstable aci–nitro compounds believed to be the substrate of glutathione-S-transferases that introduce the shared sulfur atom. The SUR1 cysteine-sulfur lyase converts the S-alkyl-thiohydroximate to thiohydroximic acid, which is then modified with a glucosyl-residue by UDP-glucosyltransferase (UGT) family 74 enzymes. The final step of the pathway involves sulfation by PAPS-dependent sulfotransferases (SOT). Subsequent modification reactions of the core structure yield the molecular diversity of glucosinolates.
For aliphatic glucosinolate synthesis, the chain-elongation process begins with deamination of methionine to the corresponding 2-oxo acid, which is catalyzed by branched-chain amino acid aminotransferase 4 (BCAT4) (
Figure 2) [
28,
29]. Although BCAT4 is localized in the cytosol, the rest of the enzymes involved in the elongation pathway are localized in the chloroplast [
30,
31,
32,
33,
34]. This indicates the need for import of 2-oxo acids into the chloroplast, a function that has been potentially assigned to a chloroplast-localized bile acid transporter BAT5 [
35]. The next reactions in the sequence require a set of three enzymes—methylthioalkylmalate synthase (MAMS), isopropylmalate isomerase (IPMI), and isopropylmalate dehydrogenase (IPMDH) (
Figure 2). These enzymes form an iterative cluster of reactions that elongate the aliphatic chain of the 2-oxo acid. MAMS catalyzes the condensation of the 2-oxo acid and acetyl-CoA to form a 2-malate derivative [
30,
31,
36,
37,
38]. Isomerization of the 2-malate derivative to a 3-malate derivative is catalyzed by IPMI [
33,
39]. The final step in the elongation cycle is performed by IPMDH, which oxidatively decarboxylates the 3-malate derivative to a 2-oxo acid, which elongates the original 2-oxo acid by a methylene group [
34,
40,
41]. After each cycle, the elongated 2-oxo acid can either proceed through another round of chain elongation or be transaminated by BCAT3 to yield homomethionine (or its elongated derivatives), which enters the core glucosinolate assembly pathway [
42,
43] (
Figure 2).
Formation of the glucosinolate core structure takes place in the cytosol and involves a set of reactions shared by aliphatic, aromatic, and indole glucosinolates (
Figure 3). Conversion of elongated methionine-derived amino acids, along with phenylalanine, tyrosine, and tryptophan, into aldoximes is performed by a set of cytochrome P450s of the CYP79 family. CYP79F1 catalyzes the reaction with all elongated methionine derivatives, while CYP79F2 only converts long-chain methionine derivatives; CYP79B3 catalyzes the reaction with tryptophan; and CYP79A2 uses phenylalanine [
44,
45,
46,
47,
48,
49]. The aldoximes are oxidized to either nitrile oxides or aci-nitro compounds by CYP83 cytochrome P450s. CYP83B1 oxidizes both the tryptophan and phenylalanine-derived acetaldoximes, while CYP83A1 converts various aliphatic aldoximes [
50,
51,
52,
53,
54].
The resulting cytochrome P450 products are conjugated to the sulfur donor glutathione by glutathione-S-transferases to produce S-alkyl-thiohydroximates, which serve as substrates for the carbon-sulfur lyase SUR1 [
55]. This is the first enzymatic transformation in the core glucosinolate biosynthesis pathway that links their assembly to enzymes of sulfur metabolism [
7,
56]. The thiohydroximates generated by SUR1 undergo S-glycosylation catalyzed by glucosyltransferases of the UGT74 family to form desulfoglucosinolates. UGT74C1 glucosylates the methionine-derived molecules and UGT74B1 modified the aromatic amino acid-derived compounds [
57,
58].
Sulfation of the desulfoglucosinolates to yield the final glucosinoate molecule is mediated by a set of sulfotransferases (SOTs) that use 3′-phosphoadenosine-5′-phosphosulfate (PAPS) as a sulfate donor [
59,
60,
61,
62,
63]. This is the second step in the assembly of glucosinolates that depends on sulfur metabolism, as the activity of the adenosine-5′-phosphosulfate kinase is essential for PAPS formation and generation of a wide array of sulfonated metabolites and that PAPS synthesis responds to sulfur nutrient levels in plants [
64,
65,
66,
67,
68,
69,
70,
71]. Like many of the enzyme families involved in glucosinolate biosynthesis, plants encode multiple SOTs with certain members of the SOT family, such as SOT16, SOT17, and SOT18 in
A. thaliana, catalyzing the modification of desulfoglucosinolates [
60,
61,
62]. Recent biochemical and X-ray crystallographic studies of Arabidopsis SOT18 identified the active site responsible for the reaction chemistry and key residues required for binding of PAPS; these features are conserved across SOT from plants and other organisms, including humans [
72]. Interestingly, the molecular basis of substrate preference of the SOT in glucosinolate biosynthesis remains elusive. For example, comparison of the residues in the SOT active site between Arabidopsis SOT16, which prefers indole desulfoglucosinolates and the methionine-derived desulfoglucosinolate metabolizing SOT17 and SOT18, suggested that the basis of substrate selectivity involves residues beyond the ligand binding site and may be dictated by multiple conformationally flexible loops near the active site [
72]. This is a biochemical problem not limited to the plant SOT, but is relevant for SOT families in other organisms, including humans.
Although the various biosynthetic steps in the aliphatic chain-elongation process and the assembly of the core glucosinolate scaffold have been determined, the molecular details for the evolution of these specialized processes and the biochemical selectivity for glucosinolate biosynthesis remain to be determined. Bioengineering efforts for the application of glucosinolates in plant defense, agriculture, human health, and animal nutrition require the knowledge of not only how glucosinolates are synthesized, but also what the specific enzymatic mechanisms and structures of enzymes involved in the glucosinolate biosynthetic pathway are.