Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Dentistry, Oral Surgery & Medicine
Periodontal inflammation is a common inflammatory disease associated with chronic inflammation that can ultimately lead to alveolar attachment loss and bone destruction. Understanding autophagy and pyroptosis has suggested their significant roles in inflammation. In recent years, studies of differentiated embryo-chondrocyte expressed genes 1 and 2 (Dec1 and Dec2) have shown that they play important functions in autophagy and in pyroptosis, which contribute to the onset of periodontal inflammation.
- Dec1
- Dec2
- periodontal inflammation
- autophagy
- pyroptosis
1. Introduction
Periodontal inflammation is a prevalent oral disease characterized by chronic inflammation caused by immune responses against various types of gram-negative anaerobic bacteria, such as P. gingivalis, T. forsythia, T. denticola, and A. actinomycetemcomitans [1]. A periodontal homeostasis imbalance in host–bacteria interactions leads to the initiation and progression of periodontal inflammation, which is regulated by many factors [2]. Pathogen infections trigger host inflammatory responses, such as the release of pro-inflammatory factors in the periodontium [3]. Chronic inflammation ultimately results in gingival recession, bone resorption, and the loss of teeth [4].
The periodontium is composed of the alveolar bone, the cementum, and the periodontal ligament (PDL), which connects the cementum to the alveolar bone [5]. The PDL contains different types of cells and serves a variety of functions. Human periodontal ligament fibroblasts (HPDLFs) constitute the majority of cells in the PDL that control alveolar bone remodeling, periodontal ligament regeneration, and protect against microbes [6]. HPDLFs can produce inflammatory mediators upon pathogenic infections, which attract immune cells including neutrophils, T lymphocytes, B lymphocytes, and macrophages that initiate inflammatory responses. Those reactions can further lead to the destruction of collagen and the resorption of alveolar bone [7].
The circadian rhythm contributes to the immune system. Circulating immune cells, pro-inflammatory cytokines, and hormones often display certain rhythms during the day [8]. Dysregulation of the circadian rhythm can disrupt the innate immune system and normal inflammatory responses [9]. Indeed, symptoms of diseases, such as rheumatoid arthritis (RA), myocardial infarction, and ischemic stroke, manifest in a circadian pattern [10,11]. The circadian rhythm is controlled by a series of transcription factors that regulate endogenous oscillations in organisms. The clock genes Dec1 and Dec2 are two members of basic helix-loop-helix (bHLH) transcription factor family and they have a 97% similarity in their bHLH region and a 52% similarity in the total proteins [12]. Dec1 and Dec2 are involved in regulating various physiological and pathological processes including the circadian rhythm, hypoxia, cell proliferation, inflammation, epithelial-to-mesenchymal transition (EMT), and carcinogenesis [13]. The expression of Dec1 and Dec2 is highly responsive to different stimuli [14,15]. Dec1 and Dec2 can be regulated by circadian rhythm proteins, such as ARNTL/CLOCK heterodimers [16,17], and they form a complex control loop in the regulation of the circadian rhythm. HIF-α directly targets the genes encoding Dec1 and Dec2 and induces the expression of Dec1 and Dec2, which is a vital process for cells adapting to hypoxia [18].
2. Autophagy
Autophagy is a lysosome-dependent degradation pathway that serves a protective role in eukaryotic cells to maintain homeostasis in response to environmental stimuli [24]. Depending on the stimulus and type of stress, autophagy can either block apoptosis or induce apoptosis [25]. Autophagy has multiple functions in cells related to cell viability, tissue damage, and the release of proinflammatory cytokines [26]. Dysregulation of autophagy has been demonstrated in the pathogenesis of inflammatory diseases including periodontal inflammation [27]. In peripheral mononuclear cells from patients with periodontal inflammation, the expression of autophagy-related protein 12 (ATG) and microtubule-associated protein 1 light chain 3 alpha (LC3) was increased. Levels of autophagy markers were also upregulated in periodontal inflammation [28] and in an experimental model of periodontal inflammation [29]. Increased autophagy in periodontal ligament stem cells (PDLSCs) was observed [30]. HPDLFs were protected from inflammation-induced apoptosis by autophagy [31]. On the other hand, in conditions of excessive stress, autophagy promotes apoptosis and inhibits cell growth [32].
Autophagy activity has been reported to appear in certain rhythm oscillations. In rat hepatocytes and cardiomyocytes, autophagy declines at feeding time and increases when at rest phase [33]. In the mouse liver, CCAAT enhancer binding protein beta (C/EBPb) is a key transcription factor that controls the diurnal autophagy rhythm [34]. Knockout of the core clock gene, circadian locomotor output cycles kaput (CLOCK), elevated the level of inflammation in mice [35].
Periodontal inflammation is characterized by a dysbiosis of oral microbiota which causes a chronic inflammatory condition [36]. Dysbiosis is characterized by the changes in the structure of the microbial species associated with diseases [37]. The augmentation of versatile polymicrobial communities were apparent in periodontal disease, whereas they were substantially decreased in healthy subjects [38,39]. Pathogens including P. gingivalis and its toxins, such as lipopolysaccharide (LPS), promote the release of inflammatory mediators, including TNF-α, IL-1β, and IL-6. Those cytokines cause the destruction of collagen and the resorption of alveolar bone in the progression of periodontal inflammation [7]. Treatment with LPS increased the number of T lymphocytes in gingival mononuclear cells [23], and it has been confirmed that LPS induces inflammation through autophagy [37].
2.1. Dec1 and Autophagy
Recent studies have demonstrated that the mammalian target of rapamycin (mTOR) is a key factor in the regulation of autophagy [38]. Autophagy can be inhibited by stimulation of the PI3K/Akt/mTOR pathway [39] or can be stimulated when the mTOR pathway is inhibited [40].
Oka et al. has recently found that P. gingivalis LPS upregulates autophagy and results in inflammation in HPDLFs. Accompanied by an elevation of IL-1β and other autophagy markers, levels of Dec1 were also significantly elevated. To further investigate the role of Dec1 in LPS induced autophagy, the effects of inhibiting Dec1 were characterized. A Dec1 deficiency affected cell autophagy levels both in vitro and in vivo and that effect was mediated via the Akt/mTOR pathway. A Dec1 deficiency could partly attenuate the inflammation level triggered by LPS-induced autophagy in mice. Those results showed the important role of Dec1 in regulating inflammation and autophagy [37].
2.2. Dec2 and Autophagy
After LPS stimulation, the ratio of LC3-II/LC3-1 was increased, indicating the upregulation of autophagy in human gingival fibroblasts [41]. The Raf/MEK/ERK signaling pathway regulates autophagy and, upon activation, LC3-II protein expression is also increased which in turn promotes autophagy [42].
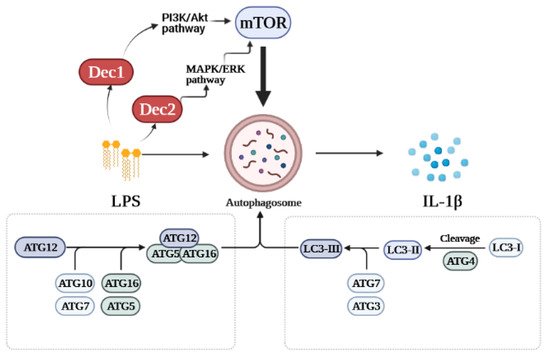
Recent in vitro and in vivo studies demonstrated that Dec2 is involved in regulating LPS-induced autophagy in HPDLFs. Knockdown of Dec2 increased autophagy and IL-1β levels in HPDLFs treated with P. gingivalis LPS although functional bindings sites were not detected between Dec2 and IL-1β. We further determined the mechanism by which Dec2 mediates autophagy. Treatment with a Dec2 siRNA indicated that Dec2 is importantly related to activation of the ERK signaling pathway, and Dec2 also inhibited autophagy via mTOR phosphorylation and subsequently 4EBP1 activation. The suppression of autophagy by Dec2 was also confirmed in an animal periodontal inflammation model [43]. Figure 1 shows a schematic diagram of the roles of Dec1 and Dec2 in autophagy. These data provide a new perspective on the pathogenesis of periodontal inflammation.
Figure 1. Schematic diagram of the roles of Dec1 and Dec2 in autophagy. Treatment with LPS stimulates the expression of Dec1 and Dec2. Dec1 mediates autophagy via the PI3K/Akt/mTOR pathway, while Dec2 regulates autophagy through the MAPK/ERK/mTOR pathway.
3. Pyroptosis
Pyroptosis is another important pathway in periodontal inflammation [44]. Pyroptosis is a distinct type of programmed cell death mediated by inflammatory caspases (caspase-1, -4, -5 and -11) and gasdermin D (GSDMD) [45]. LPS is sensed by Toll-like receptor 4 and murine caspase-11 or human caspase 4/5 [46]. The activation of caspases cleaved GSDMD, leading to pyroptosis and NOD-like receptor family and pyrin domain-containing protein 3 (NLRP3) mediated formation of inflammasomes and the release of IL-1β [45]. Caspase-1 cleaves pro-IL-1β upon activation, and IL-1β attracts immune cells causing inflammation [47]. The dysregulation of pyroptosis causes an aberrant inflammatory response, further damages the tissue, and results in inflammatory disease [47]. NF-κB contributes important functions to the progression of pyroptosis [48]. TLR4/NF-κB signaling also triggers pyroptosis [49].
NLRP3 mRNA levels display a circadian rhythm in mouse macrophages as well as the secretion of IL-1β and IL-18 [50]. A core clock component, Nuclear Receptor Subfamily 1 Group D (NR1D1), also affects IL-1β and IL-18 mRNA levels [50]. The inhibition of NR1D1 aggravates the acute lung injury and inflammation induced by LPS via the NLRP3-dependent secretion of IL-1β [51].
3.1. Dec1 and Pyroptosis
A previous study demonstrated that Dec1 can upregulate IL-1β in human gingival cells via the PI3K/AKT pathway and, thus, can influence the process of inflammation [22]. Recent studies took advantage of Dec1 KO mice to gain more insight between Dec1 and pyroptosis. Caspase-1, as well as phosphorylated NF-κB, was highly activated after LPS stimulation, indicating that pyroptosis was successfully induced. The mRNA and protein levels of IL-1β were also upregulated. Interestingly, the expression of Dec1 was significantly elevated by LPS. Dec1 deficiencies in vitro and in vivo downregulated the expression of IL-1β and NF-κB, indicating reduced pyroptosis after LPS treatment [52]. Those results suggest that Dec1 has a therapeutic potential to treat periodontal inflammation as a target in inflammation pathways.
3.2. Dec2 and Pyroptosis
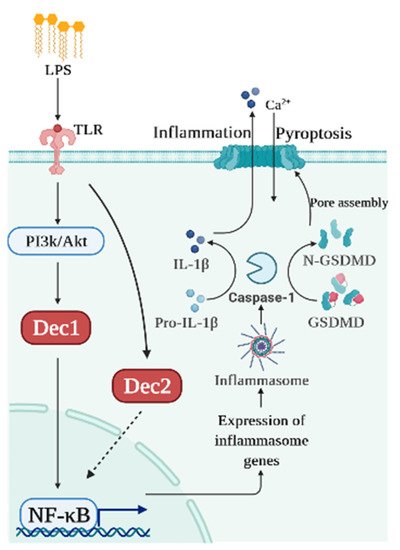
Dec2 also plays an important role in immune regulation. To explore the association between Dec2 and pyroptosis, HGFs, HPDLFs, and an experimental periodontal inflammation mouse model were studied. Cells treated with 10 μg/mL of P. gingivalis LPS showed a significantly increased expression of Dec2 and IL-1β. Caspase-1 and GSDMD were subsequently cleaved, and NF-κB was phosphorylated and then translocated into the nuclei. Both Dec2 siRNA treatment of cells and a Dec2 KO mouse periodontal inflammation model showed a regulatory function against the cleavage of GSDMD, demonstrating that Dec2 has a critical role in pyroptosis [53]. Figure 2 shows a schematic diagram summarizing the roles of Dec1 and Dec2 in pyroptosis.
Figure 2. Schematic diagram of the roles of Dec1 and Dec2 in pyroptosis. Bacterial products like LPS induce Dec1 expression and Dec1 upregulates IL-1β and NF-κB. Dec2 attenuates the phosphorylation of NF-κB which, in turn, deactivates caspase-1 and GSDMD.
This entry is adapted from the peer-reviewed paper 10.3390/ijms221910349
This entry is offline, you can click here to edit this entry!