Blastic plasmacytoid dendritic cell neoplasm (BPDCN) is an extremely rare tumour, which usually affects elderly males and presents in the skin with frequent involvement of the bone-marrow, peripheral blood and lymph nodes. It has a dismal prognosis, with most patients dying within one year when treated by conventional chemotherapies. The diagnosis is challenging, since neoplastic cells can resemble lymphoblasts or small immunoblasts, and require the use of a large panel of antibodies, including those against CD4, CD56, CD123, CD303, TCL1, and TCF4. The morphologic and in part phenotypic ambiguity explains the uncertainties as to the histogenesis of the neoplasm that led to the use of various denominations.
- blastic plasmacytoid dendritic cell neoplasm
- clinics
- morphology
- phenotype
- gene expression profile
- mutational landscape
- chemotherapy
- targeted therapy
1. Definition
2. Clinics
The disease is likely to involve multiple organs [1] with a predilection for the skin (in 60–100% cases), the bone-marrow and peripheral blood (in 60–90% of cases) and lymph nodes (in 40–50% of cases). However, the skin is the first site of presentation [3][4][5][6][7][8] (90% of patients) in the form of an isolated lesion or a rash. On close inspection, the lesions can vary from purpuric papules (often misdiagnosed as petechiae) to violaceous patches, plaques or tumors, often resembling insect bites, malignant lymphoma (such as mycosis fungoides or Sezary syndrome) or Kaposi sarcoma. Another possible differential diagnosis can be drug eruption. In such cases a careful clinical history and inspection as well as a careful clinical inspection is mandatory to avoid misdiagnosis. Usually, BPDCN does not show a preferred anatomical area and therefore may virtually affect any site of the body. Unfortunately, the disease remains confined to skin for a variably, yet generally short time, and then the patient starts to complain B-symptoms (i.e. night sweat, fever, fatigue and malaise) due to the dissemination of the disease to internal organs or to bone marrow and blood. Such a second step is usually rapid and precedes the death of the patient. Due to its common and very first involvement, it has been hypothesized that the skin may limit disease spread at the beginning [9] and therefore should be considered as a sanctuary organ. However, a few cases lacking the initial cutaneous involvement have been reported [10][11][12][13][14]. A possible explanation of the BPDCN cutaneous tropism may rely on the expression of skin-migration molecules such as CLA and CD56 by the neoplastic elements as well as the expression of chemokines binding cognate receptor such as CXCR3, CXCR4, CXCR6, CXCR7. In the skin, the disease can present as isolated or disseminated lesions [7][15]. On clinical grounds, the distinction between isolated and eruptive lesions [9] may be important, owing to the report in the literature of patients with a single lesion featuring a better clinical outcome than those showing multiple lesions [9]. A possible explanation may be due to a different tumor burden in the two presentations. Cases featuring mucosal involvement, especially in the oral cavity have rarely been observed [8].
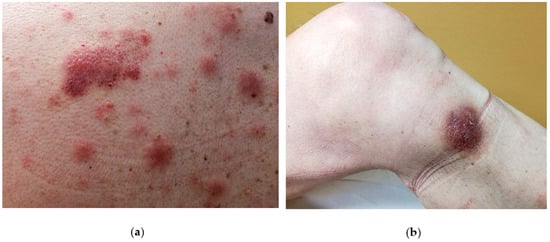
3. Molecular features of Blastic Plasmacytoid Dendritic Neoplasm
3.1 Karyotyping
BPDCN patients are affected by frequent chromosomal alterations: up to 75% of them present a complex karyotype (≥3 abnormalities). Conventional cytogenetics studies reported a prevalence of genomic losses on gains and recognized six recurrent deletions of the regions 5q21 or 5q34 (72%), 12p13 (64%), 13q13-21 (64%), 6q23-qter (50%), 15q (43%) and of the entire chromosome 9 (28%) [16][17]. Although recurrent, none of these alterations turned out to be BPDCN-specific, also being observed in other hematological malignancies.
By FISH analysis, MYC translocations were reported in the 39% of BPDCN patients, in association with the above mentioned immunoblast-like morphology [18]. T(6;8)(p21;q24) corresponded to the commonest type of MYC rearrangement: it defined a subgroup of patients with a more aggressive behavior [19]. Of clinical relevance, MYC positivity was found to confer good response to the acute lymphoblastic leukemia (ALL)–based chemotherapy in a limited number of patients [18][20]. Furthermore, FISH analysis documented in a few cases the translocation of the MLL1 gene, also recorded in 18% of ALLs [21][22][23][24], along with frequent rearrangements of the ETS variant gene 6 (ETV6), a transcription factor disrupted in other hematological malignancies [25][26].
Akin to cytogenetics and FISH, a-CGH evidenced frequent deletions on the chromosomes 4, 9 and 13. Lucioni et al. analyzed by a-CGH 21 BPDCNs and found that the most affected chromosomes were Chr 9 (71%), 13 (61%), 12 (57%), 5 (19%), 7 (19%), 14 (19%), and 15 (14%). The deletions outnumbered the amplifications and resulted in the loss of CDKN1B, CDKN2A, CDKN2B, RB1, LATS2, and IKZF1 [27]. While aberrations of IKZF1, which is involved in the regulation of dendritic cell hematopoiesis, may cause pDCs deficiency [28], their impact on BPDCN (see below) remains indeterminate yet. The biallelic deletion of CDKN2A predicted a worse survival outcome [27]. Wiesner et al. performed a-CGH and immunostaining analysis of 14 BPDCN skin samples and confirmed recurrent deletions along chromosomes 9, 12, 13, and 15, combined with the negative or weak expression of multiple cell-cycle and tumor suppressor genes (e.g., CDKN1B, CDKN2A, RB1, and TP53) [29], possibly responsible for the uncontrolled proliferation and aggressiveness of BPDCN tumor cells [29][30].
3.2 Gene Expression Profiling by Array
The first study of BPDCN gene expression profiling (GEP) was conducted in 2007 by Dijkman et al. Since BPDCN skin lesions could easily be confused with cutaneous myelomonocytic leukemia (c-AML), Dijkman et al. performed a-CGH and GEP by array of 5 BPDCN skin biopsies and 6 c-AML cases. According to their study, BPDCN displayed: (1) a transcriptome profile and a molecular karyotype indeed distinct from c-AMLs; (2) recurrent deletions of 4q34, 9, and 13q12-q31 chromosomal regions; (3) lower expression of RB1 and LATS2 tumor suppressor genes; (4) higher expression of various pDC-related genes, such as the TLRs, TLR9 and TLR10 [31].
In 2014, Sapienza et al. compared for the first time the gene signature of 27 BPDCN primary samples with that of normal pDCs and found that the tumor transcriptome was more similar to resting pDCs rather than activated ones, confirming at molecular level the origin of BPDCN from a pDC precursor [2]. Tumor samples displayed 142 differentially expressed genes, mostly upregulated (89%), including those encoding for CyclinD1 and the anti-apoptotic protein BCL2. Bioinformatic analysis of GEP data revealed the aberrant activation of the NF-kB pathway, a finding suggesting possible response of BPDCN samples/cell lines to the proteasome inhibitor Bortezomib [2]. In vitro and in vivo experiments demonstrated that Bortezomib successfully shuts-down the NF-kB pathway and significantly induces BPDCN cell apoptosis, providing a potential new therapeutic option for BPDCN patients [2][32].
Ceroi et al. performed transcriptional profiling of 12 BPDCN cases by array and focused on a specific signature of downregulated genes involved in cholesterol homeostasis and responsible for its accumulation within the tumor cells. These sets of downregulated genes, if activated, stimulated the cholesterol efflux from neoplastic cells, inhibited the NF-kB pathway and arrested the BPDCN tumor cell survival [33].
3.3 DNA Sequencing and Next Generation Sequencing
The chromosomal lesions of BPDCNs fully reflect their myeloid origin and the same could be expected at the DNA mutational level. Starting from the premise that the mutations of the epigenetic regulator gene TET2 are commonly observed in the myeloid lineage [34], Jardin et al. decided to explore the mutational status of this gene in 13 BPDCNs. TET2 was mutated in more than half of patients and was mostly affected by deleterious mutations (frameshift or nonsense). At diagnosis, TET2 mutations (54%) were recurrently flanked by TP53 mutations (38%) leading to hypothesize a synergistic effect between the two genes [35]. Alayed et al. confirmed the high mutational frequency of TET2 in BPDCN [36]. Ladikou et al. conducted the first targeted-sequencing on the BPDCN circulating free DNA of BPDCN cases, by identifying novel mutations of TET2 and RHOA [37]. Besides TET2, thanks to the targeted sequencing approach, many other myeloid-associated genes have been investigated in BPDCN. Taylor et al. presented to the ASH Meeting the first study of targeted sequencing of 219 myeloid-related genes in seven BPDCN samples. The most frequently mutated gene was the splicing factor ZRSR2 (57%) ex aequo with TET2 (57%), followed by ASXL1, TP53 and IDH2, KRAS, ABL1, ARID1A, GNA13, U2AF1, SRSF2, and the transcription factor IRF8 associated with dendritic cell deficiency [38]. Later, Stenzinger et al. sequenced 50 common myeloid genes in 33 cases of BPDCN and, in order of prevalence, detected somatic mutations on NRAS, ATM, KRAS, MET, IDH2, KIT, RB1, APC, TP53, RET, VHL, BRAF, and MLH1 genes, and deletions of CDKN2A, RB1, PTEN, and TP53 genes, already found by a-CGH [39]. Menezes et al. analyzed three patients by whole exome sequencing (WES) and used the WES results to design a targeted-sequencing panel of selected genes to examine 38 BPDCN samples. The most affected genes were TET2 (36%), ASXL1 (32%), NRAS, NPM1, and IKZF family 1/2/3 (20%). Overall, 50% and 20% of patients with mutations in genes encoding for epigenetic factors or belonging to the IKAROS family respectively experienced a significantly reduced overall survival [40].
More recently, integrated “omics” approaches have been applied aiming to better understand the tumor biology. Montero et al. analyzed, by RNA-sequencing, 12 BPDCN samples and four pDCs from healthy donors by confirming BCL2 overexpression in tumors. Furthermore, by the BH3-proling of two BPDCN cell lines (CAL-1 and GEN2.2), six primary patient samples, and six patient-derived xenografts, the same authors demonstrated the BCL2 dependence of BPDCN elements as well as their sensitivity to the BCL2 inhibitor venetoclax. In the light of this finding, two patients were then treated with venetoclax and experienced significant disease responses [41][42].
Ceribelli et al. first performed an RNA interference screening study of the CAL-1 BPDCN cell line and recognized the transcription factor TCF4 as a master regulator of the BPDCN oncogenic program: its downregulation provoked the loss of the BPDCN-specific gene expression signature along with tumor cell death. Already described as relevant in normal pDC development, the TCF4 gene product was positively detected by immunohistochemistry in all the 28 BPDCN samples examined and proposed as a new reliable diagnostic marker (see above) and potential therapeutic target for bromodomain and extra-terminal domain inhibitors (BETis) [43].
Emadali et al. further substantiated the use of BETis in BPDCN. They examined 47 tumor samples and the CAL-1 cell line by various techniques (e.g., cytogenetics, a-CGH, FISH, targeted sequencing) and found that the loss of the glucocorticoid receptor gene, NR3C1, defined a high-risk group of patients. NR3C1 is often juxtaposed with lncRNA3q, a novel nuclear noncoding RNA involved in the regulation of leukemia stem cell programs and G1/S transition and aberrantly overexpressed in BPDCN malignant cells. BETis successfully turned-off the expression of lncRNA3q and inhibited tumor cells growth [44].
Suzuki et al., used RNA sequencing technology to discover novel fusion genes in 14 BPDCNs corresponding to five children and nine adults; recurrent MYB gene rearrangement were identified in all the children (100%) and in four out of the nine adults (44%) [45].
Sapienza et al. analyzed BPDCNs by WES, RNA and Chromatin Immunoprecipitation (ChIP) sequencing approaches. Several epigenetic factor genes were found mutated (e.g., ASXL1, TET2, SUZ12, ARID1A, PHF2, CHD8) and the functional enrichment analysis of the mutational data showed that of all the biological programs explored, the epigenetic was the most affected. At transcriptomic level, the patients displayed the significant enrichment of gene signatures related to epigenetic pathways, predicting response to hypomethylating agents. Accordingly, the use of 5’-azacytidine in combination with decitabine significantly inhibited disease progression and extended survival in a preclinical mouse model [46].
Bastidas Torres et al. published the first work of whole genome sequencing of 10 BPDCN cases, 4 of them also analyzed at transcriptomic level by RNA sequencing. BPDCN displayed a heterogenous landscape of genomic alterations. Most of genes disrupted by rearrangements were involved in cytoskeleton‐associated processes, adhesion and transcriptional regulation. Of pathobiological interest, IKZF1 gene was recurrently affected by deletions or unbalanced rearrangements. According to the authors, since IKZF1 is critical for pDC differentiation, its deficiency in BPDCN may derail the normal development of pDC precursors, possibly leading to their malignant transformation [47].
The cellular origin of BPDCN has been debated for years, still PDCN has been finally recognized as a neoplasm deriving from pDC in resting state [2]. However, a new pDC population has been recently recognized, AXL and SIGLEC6 positive, called AS-DCs [48]. These cells secrete low interferon, are able to stimulate T-cell proliferation and are supposed to be a transition state from plasmacytoid to myeloid dendritic cells. Renosi et al conducted a study of gene expression profile and target sequencing to clarify whether BPDCN derive from canonical pDCs or from AS-DCs. BPDCN cases displayed profiles evoking either the classical pDC or AS-DC signatures. Overall, this study highlighted the heterogeneity of BPDCN and its difficult diagnosis [49].
Sapienza et al in 2020 analyzed the microRNA expression profiling of BPDCN respect to myeloid sarcoma (MS) and identified a set of 12 miRNAs useful to discriminate between BPDCN and MS in cases with defective/ambiguous phenotype [50]. Later the same research group, integrated microRNA and gene expression profile data and developed the first BPDCN miRNA regulatory network. The primary function of miRNA network resulted to be the modulation of neurogenesis, a biological process usually referred to neuronal cells. However, more than 30% of BPDCN patients relapse in the central nervous system where BPDCN tumor cells interact with nervous elements. The neurogenesis was investigated in BPDCN by querying different molecular data (RNA sequencing, Chromatin immunoprecipitation-sequencing, and immunohistochemistry) and BPDCN cells showed to upregulate neural mitogen genes possibly critical for tumor dissemination and cross-talk with the nervous system [51].
4. Therapy of Blastic Plasmacytoid Dendritic Neoplasm
BPDCN is characterized by an inherent resistance to standard chemotherapies. Treatment responses are mostly transient, the overall outcome being general very poor in general [12][52][53]. Given the rarity of the disease, the available data on BPDCN therapy mainly derive from retrospective studies.
In general, intensive induction regimens (e.g., hyperCVAD) are considered more effective compared to standard therapies (e.g., CHOP-like) [53][54][55]. In general, ALL-like treatments seem to be more effective in term of response rates than AML-like induction therapies [12][52][53]. The inclusion of l-asparaginase in ALL-like regimens could be a significant determinant of efficacy in this setting, as l-asparaginase has shown clinical activity in BPDCN in combination with single agent methotrexate [56][57].
Regarding the role of hematopoietic stem cell transplant in BPDCN therapy, there are several reports suggesting better results in terms of enduring remissions and relapse rates with allogeneic-stem cell transplantation (allo-SCT) compared to auto-SCT. These studies demonstrated durable complete remissions with allo-SCT, with OS rates ranging from 40% at 10 years, to 58% at 3 years depending on the follow-up period [58][59]. In general, allo-SCT consolidation seems to yield the best results when performed in first complete remission (CR) [59][60][61], with OS rates reaching 74–82% at 3–4 years [59][61]. Reduced intensity conditioning seems to be equivalent to myeloablative regimens in terms of relapse rates [59]. However, these data should be interpreted with due caution given the possible biases arising from the retrospective nature of these studies (e.g., patient selection bias, absence of intention to treat analyses, small sample size).
Eligible patients should be considered for allo-SCT consolidation in first CR whenever feasible. It should be noted, however, that these patients represent the minority of BPDCN patients, as the disease normally affects elderly patients, with a median age of 68 years [12].
For elderly patients, lower intensity treatments can be explored. Lower intensity chemotherapy regimens demonstrated some efficacy in BPDCN, such as single agent pralatrexate, bendamustine, or gemcitabine/docetaxel combinations [62][63][64][65]. However, despite the promising results, these studies were performed on a small number of patients, and which ought to be validated in larger future studies.
A recent study by our group strongly supports the use of hypomethylating agents, demonstrating a significant enrichment in epigenetic modifiers mutations in the setting of BPDCN [46]. In line with our preclinical findings, two clinical reports have demonstrated activity of 5-azacitidine in BPDCN, although the responses were generally transient once again [66][67]. Combinatory approaches based on hypomethylating agents should be explored in the near future. In patients older than 70 years with co-morbidities limited-field or total-skin radiotherapeutic protocols should be considered as palliative treatment [9] if the disease is still confined to the skin.
Novel Agents
Given the unsatisfactory results of low-intensity treatments, and the toxicity of intensive therapies and allo-SCT consolidation, there is strong rationale for the use of novel targeted agents for the treatment of BPDCN.
SL-401 is a novel recombinant protein including components of diphtheria toxin fused to interleukin-3. As mentioned in previous sections, CD123 is expressed on the surface of BPDCN cells. In a phase I study of SL-401 in BPDCN the overall response rate was 77% (with 55% CR) in the evaluable patient population (seven out of eleven patients were able to complete the planned treatment) [68]. A phase 2 study reported at the 2017 ASH meeting showed promising results with a 79% CR rate in first line and 31% CR rate in relapsed/refractory patients [69].
Phase I trials are ongoing with other immunotherapies targeting CD123, such as bispecific antibodies, immunoconjugates, and chimeric antigen receptor (CAR)-T-cells [70]. In fact, recent data show promising activity of anti-CD123 CAR T-cells in acute myeloid leukemia and preliminary experiences support the future implementation of anti-CD123 CAR-T cell therapy in the BPDCN setting [71]. The BCL-2 inhibitor venetoclax has shown high single agent activity in myeloid malignancies [72][73] and is currently under evaluation in combination with induction chemotherapy and hypomethylating agents.
Several recently published reports have described the activity of venetoclax in the setting of BPDCN [42][73][74][75]. Venetoclax given as single agent or in combination with hypomethylating agents was able to induce meaningful clinical responses in relapsed/refractory patients. Further therapeutic options may be represented by bromodomain and extra-terminal domain inhibitors (BETis), which has been tested in preclinical studies [43][44].
This entry is adapted from the peer-reviewed paper 10.3390/cancers11050595
References
- Swerdlow S.H., Campo E., Harris N.L., Jaffe E.S., Pileri S.A., Stein H., Thiele J., editors. World Health Organization Classification of Tumours.4th ed. International Agency for Research on Cancer; Lyon, France: 2017. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.
- Sapienza M.R., Fuligni F., Agostinelli C., Tripodo C., Righi S., Laginestra M.A., Pileri A., Mancini M., Rossi M., Ricci F., et al. Molecular profiling of blastic plasmacytoid dendritic cell neoplasm reveals a unique pattern and suggests selective sensitivity to NF-kB pathway inhibition. Leukemia. 2014;28:1606–1616. doi: 10.1038/leu.2014.64. [PMC free article]
- Bekkenk M.W., Jansen P.M., Meijer C.J.L.M., Willemze R. CD56+ hematological neoplasms presenting in the skin: A retrospective analysis of 23 new cases and 130 cases from the literature. Ann. Oncol. 2004;15:1097–1108. doi: 10.1093/annonc/mdh268.
- Petrella T., Meijer C.J.L.M., Dalac S., Willemze R., Maynadié M., Machet L., Casasnovas O., Vergier B., Teitell M.A. TCL1 and CLA expression in agranular CD4/CD56 hematodermic neoplasms (blastic NK-cell lymphomas) and leukemia cutis. Am. J. Clin. Pathol. 2004;122:307–313. doi: 10.1309/0QPPAVTUPCV9UCLV.
- Suzuki R., Nakamura S., Suzumiya J., Ichimura K., Ichikawa M., Ogata K., Kura Y., Aikawa K., Teshima H., Sako M., et al. Blastic natural killer cell lymphoma/leukemia (CD56-positive blastic tumor): Prognostication and categorization according to anatomic sites of involvement. Cancer. 2005;104:1022–1031. doi: 10.1002/cncr.21268.
- Assaf C., Gellrich S., Whittaker S., Robson A., Cerroni L., Massone C., Kerl H., Rose C., Chott A., Chimenti S., et al. CD56-positive haematological neoplasms of the skin: A multicentre study of the Cutaneous Lymphoma Project Group of the European Organisation for Research and Treatment of Cancer. J. Clin. Pathol. 2007;60:981–989. doi: 10.1136/jcp.2006.042135. [PMC free article]
- Cota C., Vale E., Viana I., Requena L., Ferrara G., Anemona L., Metze D., Fink-Puches R., Wiesner T., Cerroni L. Cutaneous manifestations of blastic plasmacytoid dendritic cell neoplasm-morphologic and phenotypic variability in a series of 33 patients. Am. J. Surg. Pathol. 2010;34:75–87. doi: 10.1097/PAS.0b013e3181c5e26b.
- Julia F., Petrella T., Beylot-Barry M., Bagot M., Lipsker D., Machet L., Joly P., Dereure O., Wetterwald M., d’Incan M., et al. Blastic plasmacytoid dendritic cell neoplasm: Clinical features in 90 patients. Br. J. Dermatol. 2013;169:579–586. doi: 10.1111/bjd.12412.
- Pileri A., Delfino C., Grandi V., Agostinelli C., Pileri S.A., Pimpinelli N. Blastic plasmacytoid dendritic cell neoplasm (BPDCN): The cutaneous sanctuary. G. Ital. Dermatol. Venereol. 2012;147:603–608.
- Rauh M.J., Rahman F., Good D., Silverman J., Brennan M.K., Dimov N., Liesveld J., Ryan D.H., Burack W.R., Bennett J.M. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation, lacking cutaneous involvement: Case series and literature review. Leuk. Res. 2012;36:81–86. doi: 10.1016/j.leukres.2011.07.033.
- Wang H., Cao J., Hong X. Blastic plasmacytoid dendritic cell neoplasm without cutaneous lesion at presentation: Case report and literature review. Acta Haematol. 2012;127:124–127. doi: 10.1159/000334703.
- Pagano L., Valentini C.G., Pulsoni A., Fisogni S., Carluccio P., Mannelli F., Lunghi M., Pica G., Onida F., Cattaneo C., et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: An Italian multicenter study. Haematologica. 2013;98:239–246. doi: 10.3324/haematol.2012.072645. [PMC free article]
- Endo K., Mihara K., Oiwa H., Yoshida T., Mino T., Sasaki N., Takihara Y. Lung involvement at initial presentation in blastic plasmacytoid dendritic cell neoplasm lacking cutaneous lesion. Ann. Hematol. 2013;92:269–270. doi: 10.1007/s00277-012-1557-4.
- Paluri R., Nabell L., Borak S., Peker D. Unique presentation of blastic plasmacytoid dendritic cell neoplasm: A single-center experience and literature review. Hematol. Oncol. 2015;33:206–211. doi: 10.1002/hon.2147.
- Facchetti F., Cigognetti M., Fisogni S., Rossi G., Lonardi S., Vermi W. Neoplasms derived from plasmacytoid dendritic cells. Mod. Pathol. 2016;29:98–111. doi: 10.1038/modpathol.2015.145.
- Petrella T., Dalac S., Maynadié M., Mugneret F., Thomine E., Courville P., Joly P., Lenormand B., Arnould L., Wechsler J., et al. CD4+ CD56+ Cutaneous Neoplasms: A Distinct Hematological Entity? Am. J. Surg. Pathol. 1999;23:137–146. doi: 10.1097/00000478-199902000-00001.
- Leroux D. CD4+, CD56+ DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: A study of 21 cases by the Groupe Francais de Cytogenetique Hematologique. Blood. 2002;99:4154–4159. doi: 10.1182/blood.V99.11.4154.
- Sakamoto K., Katayama R., Asaka R., Sakata S., Baba S., Nakasone H., Koike S., Tsuyama N., Dobashi A., Sasaki M., et al. Recurrent 8q24 rearrangement in blastic plasmacytoid dendritic cell neoplasm: Association with immunoblastoid cytomorphology, MYC expression, and drug response. Leukemia. 2018;32:2590–2603. doi: 10.1038/s41375-018-0154-5.
- Sumarriva Lezama L., Chisholm K.M., Carneal E., Nagy A., Cascio M.J., Yan J., Chang C.-C., Cherry A., George T.I., Ohgami R.S. An analysis of blastic plasmacytoid dendritic cell neoplasm with translocations involving the MYC locus identifies t(6;8)(p21;q24) as a recurrent cytogenetic abnormality. Histopathology. 2018;73:767–776. doi: 10.1111/his.13668.
- Boddu P.C., Wang S.A., Pemmaraju N., Tang Z., Hu S., Li S., Xu J., Medeiros L.J., Tang G. 8q24/MYC rearrangement is a recurrent cytogenetic abnormality in blastic plasmacytoid dendritic cell neoplasms. Leuk. Res. 2018;66:73–78. doi: 10.1016/j.leukres.2018.01.013.
- Leung R., Chow E.E., Au W.-Y., Chow C., Kwong Y.-L., Lin S.-Y., Ma E.S., Wan T.S., Wong K.-F. CD4+/CD56+ hematologic malignancy with rearranged MLL gene. Hum. Pathol. 2006;37:247–249. doi: 10.1016/j.humpath.2005.10.005.
- Toya T., Nishimoto N., Koya J., Nakagawa M., Nakamura F., Kandabashi K., Yamamoto G., Nannya Y., Ichikawa M., Kurokawa M. The first case of blastic plasmacytoid dendritic cell neoplasm with MLL-ENL rearrangement. Leuk. Res. 2012;36:117–118. doi: 10.1016/j.leukres.2011.07.029.
- Yang N., Huh J., Chung W.S., Cho M.-S., Ryu K.-H., Chung H.-S. KMT2A (MLL)-MLLT1 rearrangement in blastic plasmacytoid dendritic cell neoplasm. Cancer Genet. 2015;208:464–467. doi: 10.1016/j.cancergen.2015.04.011.
- Meyer C., Burmeister T., Gröger D., Tsaur G., Fechina L., Renneville A., Sutton R., Venn N.C., Emerenciano M., Pombo-de-Oliveira M.S., et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–284. doi: 10.1038/leu.2017.213. [PMC free article]
- Tang Z., Li Y., Wang W., Yin C.C., Tang G., Aung P.P., Hu S., Lu X., Toruner G.A., Medeiros L.J., et al. Genomic aberrations involving 12p/ETV6 are highly prevalent in blastic plasmacytoid dendritic cell neoplasms and might represent early clonal events. Leuk. Res. 2018;73:86–94. doi: 10.1016/j.leukres.2018.09.006.
- Haferlach C., Bacher U., Schnittger S., Alpermann T., Zenger M., Kern W., Haferlach T. ETV6 rearrangements are recurrent in myeloid malignancies and are frequently associated with other genetic events. Genes Chromosomes Cancer. 2012;51:328–337. doi: 10.1002/gcc.21918.
- Lucioni M., Novara F., Fiandrino G., Riboni R., Fanoni D., Arra M., Venegoni L., Nicola M., Dallera E., Arcaini L., et al. Twenty-one cases of blastic plasmacytoid dendritic cell neoplasm: Focus on biallelic locus 9p21.3 deletion. Blood. 2011;118:4591–4594. doi: 10.1182/blood-2011-03-337501.
- Cytlak U., Resteu A., Bogaert D., Kuehn H.S., Altmann T., Gennery A., Jackson G., Kumanovics A., Voelkerding K.V., Prader S., et al. Ikaros family zinc finger 1 regulates dendritic cell development and function in humans. Nat. Commun. 2018;9:1239. doi: 10.1038/s41467-018-02977-8. [PMC free article]
- Wiesner T., Obenauf A.C., Cota C., Fried I., Speicher M.R., Cerroni L. Alterations of the Cell-Cycle Inhibitors p27KIP1 and p16INK4a Are Frequent in Blastic Plasmacytoid Dendritic Cell Neoplasms. J. Investig. Dermatol. 2010;130:1152–1157. doi: 10.1038/jid.2009.369.
- Jardin F., Callanan M., Penther D., Ruminy P., Troussard X., Kerckaert J.P., Figeac M., Parmentier F., Rainville V., Vaida I., et al. Recurrent genomic aberrations combined with deletions of various tumour suppressor genes may deregulate the G1/S transition in CD4+CD56+ haematodermic neoplasms and contribute to the aggressiveness of the
- Dijkman R., van Doorn R., Szuhai K., Willemze R., Vermeer M.H., Tensen C.P. Gene-expression profiling and array-based CGH classify CD4+CD56+ hematodermic neoplasm and cutaneous myelomonocytic leukemia as distinct disease entities. Blood. 2007;109:1720–1727. doi: 10.1182/blood-2006-04-018143.
- Philippe L., Ceroi A., Bôle-Richard E., Jenvrin A., Biichle S., Perrin S., Limat S., Bonnefoy F., Deconinck E., Saas P., et al. Bortezomib as a new therapeutic approach for blastic plasmacytoid dendritic cell neoplasm. Haematologica. 2017;102:1861–1868. doi: 10.3324/haematol.2017.169326. [PMC free article]
- Ceroi A., Masson D., Roggy A., Roumier C., Chagué C., Gauthier T., Philippe L., Lamarthée B., Angelot-Delettre F., Bonnefoy F., et al. LXR agonist treatment of blastic plasmacytoid dendritic cell neoplasm restores cholesterol efflux and triggers apoptosis. Blood. 2016;128:2694–2707. doi: 10.1182/blood-2016-06-724807. [PMC free article]
- Mullighan C.G. TET2 mutations in myelodysplasia and myeloid malignancies. Nat. Genet. 2009;41:766–767. doi: 10.1038/ng0709-766.
- Jardin F., Ruminy P., Parmentier F., Troussard X., Vaida I., Stamatoullas A., Leprêtre S., Penther D., Duval A.B., Picquenot J.-M., et al. TET2 and TP53 mutations are frequently observed in blastic plasmacytoid dendritic cell neoplasm: Correspondence. Br. J. Haematol. 2011;153:413–416. doi: 10.1111/j.1365-2141.2010.08556.x.
- Alayed K., Patel K.P., Konoplev S., Singh R.R., Routbort M.J., Reddy N., Pemmaraju N., Zhang L., Shaikh A.A., Aladily T.N., et al. TET2 mutations, myelodysplastic features, and a distinct immunoprofile characterize blastic plasmacytoid dendritic cell neoplasm in the bone marrow. Am. J. Hematol. 2013;88:1055–1061. doi: 10.1002/ajh.23567.
- Ladikou E., Ottolini B., Nawaz N., Allchin R.L., Payne D., Ali H., Marafioti T., Shaw J., Ahearne M.J., Wagner S.D. Clonal evolution in the transition from cutaneous disease to acute leukemia suggested by liquid biopsy in blastic plasmacytoid dendritic cell neoplasm. Haematologica. 2018;103:e196–e199. doi: 10.3324/haematol.2017.171876. [PMC free article]
- Taylor J., Kim S.S., Stevenson K.E., Yoda A., Kopp N., Louissaint A., Harris N.L., Hochberg E.P., Chen Y.-B., Lovitch S.B., et al. Loss-of-function mutations in the splicing factor ZRSR2 are common in blastic Plasmacytoid Dendritic cell neoplasm and have male predominance. Blood. 2013;122:741.
- Stenzinger A., Endris V., Pfarr N., Andrulis M., Jöhrens K., Klauschen F., Siebolts U., Wolf T., Koch P.-S., Schulz M., et al. Targeted ultra-deep sequencing reveals recurrent and mutually exclusive mutations of cancer genes in blastic plasmacytoid dendritic cell neoplasm. Oncotarget. 2014;5:15. doi: 10.18632/oncotarget.2223. [PMC free article]
- Menezes J., Acquadro F., Wiseman M., Gómez-López G., Salgado R.N., Talavera-Casañas J.G., Buño I., Cervera J.V., Montes-Moreno S., Hernández-Rivas J.M., et al. Exome sequencing reveals novel and recurrent mutations with clinical impact in blastic plasmacytoid dendritic cell neoplasm. Leukemia. 2014;28:823–829. doi: 10.1038/leu.2013.283.
- Montero J., Stephansky J., Cai T., Griffin G.K., Togami K., Cabal-Hierro L., LeBoeuf N.R., Hogdal L., Galinsky I., Aster J.C., et al. Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) Is Highly BCL-2 Dependent and Sensitive to Venetoclax. Blood. 2016;128:4045. [PMC free article]
- Montero J., Stephansky J., Cai T., Griffin G.K., Cabal-Hierro L., Togami K., Hogdal L.J., Galinsky I., Morgan E.A., Aster J.C., et al. Blastic Plasmacytoid Dendritic Cell Neoplasm Is Dependent on BCL2 and Sensitive to Venetoclax. Cancer Discov. 2017;7:156–164. doi: 10.1158/2159-8290.CD-16-0999. [PMC free article]
- Ceribelli M., Hou Z.E., Kelly P.N., Huang D.W., Wright G., Ganapathi K., Evbuomwan M.O., Pittaluga S., Shaffer A.L., Marcucci G., et al. A Druggable TCF4- and BRD4-Dependent Transcriptional Network Sustains Malignancy in Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancer Cell. 2016;30:764–778. doi: 10.1016/j.ccell.2016.10.002. [PMC free article]
- Emadali A., Hoghoughi N., Duley S., Hajmirza A., Verhoeyen E., Cosset F.-L., Bertrand P., Roumier C., Roggy A., Suchaud-Martin C., et al. Haploinsufficiency for NR3C1, the gene encoding the glucocorticoid receptor, in blastic plasmacytoid dendritic cell neoplasms. Blood. 2016;127:3040–3053. doi: 10.1182/blood-2015-09-671040. [PMC free article]
- Suzuki K., Suzuki Y., Hama A., Muramatsu H., Nakatochi M., Gunji M., Ichikawa D., Hamada M., Taniguchi R., Kataoka S., et al. Recurrent MYB rearrangement in blastic plasmacytoid dendritic cell neoplasm. Leukemia. 2017;31:1629–1633. doi: 10.1038/leu.2017.101.
- Sapienza M.R., Abate F., Melle F., Orecchioni S., Fuligni F., Etebari M., Tabanelli V., Laginestra M.A., Pileri A., Motta G., et al. Blastic plasmacytoid dendritic cell neoplasm: Genomics mark epigenetic dysregulation as a primary therapeutic target. Haematologica. 2018;104:729–737. doi: 10.3324/haematol.2018.202093. [PMC free article]
- Bastidas Torres, A.N.; Cats, D.; Mei, H.; Fanoni, D.; Gliozzo, J.; Corti, L.; Paulli, M.; Vermeer, M.H.; Willemze, R.; Berti, E.; et al. Whole-Genome Analysis Uncovers Recurrent IKZF1 Inactivation and Aberrant Cell Adhesion in Blastic Plasmacytoid Dendritic Cell Neoplasm. Genes Chromosomes Cancer 2020, 59, 295–308, doi:10.1002/gcc.22831.
- Villani, A.-C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-Cell RNA-Seq Reveals New Types of Human Blood Dendritic Cells, Monocytes, and Progenitors. Science 2017, 356, eaah4573, doi:10.1126/science.aah4573.
- Renosi, F.; Roggy, A.; Giguelay, A.; Soret, L.; Viailly, P.-J.; Cheok, M.; Biichle, S.; Angelot-Delettre, F.; Asnafi, V.; Macintyre, E.; et al. Transcriptomic and Genomic Heterogeneity in Blastic Plasmacytoid Dendritic Cell Neoplasms: From Ontogeny to Oncogenesis. Blood Adv 2021, 5, 1540–1551, doi:10.1182/bloodadvances.2020003359.
- Sapienza, M.R.; Fuligni, F.; Melle, F.; Tabanelli, V.; Indio, V.; Laginestra, M.A.; Motta, G.; Mazzara, S.; Cerroni, L.; Pileri, A.; et al. MicroRNA Profiling of Blastic Plasmacytoid Dendritic Cell Neoplasm and Myeloid Sarcoma. Hematological Oncology 2020, 38, 831–833, doi:10.1002/hon.2782.
- Sapienza, M.R.; Benvenuto, G.; Ferracin, M.; Mazzara, S.; Fuligni, F.; Tripodo, C.; Belmonte, B.; Fanoni, D.; Melle, F.; Motta, G.; et al. Newly-Discovered Neural Features Expand the Pathobiological Knowledge of Blastic Plasmacytoid Dendritic Cell Neoplasm. Cancers 2021, 13, 4680, doi:10.3390/cancers13184680.
- Tsagarakis N.J., Kentrou N.A., Papadimitriou K.A., Pagoni M., Kokkini G., Papadaki H., Pappa V., Marinakis T., Anagnostopoulos N.I., Vadikolia C., et al. Acute lymphoplasmacytoid dendritic cell (DC2) leukemia: Results from the Hellenic Dendritic Cell Leukemia Study Group. Leuk. Res. 2010;34:438–446. doi: 10.1016/j.leukres.2009.09.006. [PubMed]
- Deotare U., Yee K.W.L., Le L.W., Porwit A., Tierens A., Musani R., Barth D., Torlakovic E., Schimmer A., Schuh A.C., et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: 10-Color flow cytometry diagnosis and HyperCVAD therapy: BPDCN Diagnosis and Therapy. Am. J. Hematol. 2016;91:283–286. doi: 10.1002/ajh.24258.
- Feuillard J., Jacob M.-C., Valensi F., Maynadié M., Gressin R., Chaperot L., Arnoulet C., Brignole-Baudouin F., Drénou B., Duchayne E., et al. Clinical and biologic features of CD4(+)CD56(+) malignancies. Blood. 2002;99:1556–1563. doi: 10.1182/blood.V99.5.1556.
- Deotare U., Kim D., Dong H., Michelis F.V., Lipton J.H. Allogeneic Hematopoietic Stem Cell Transplantions in Blastic Plasmacytoid Dendritic Cell Neoplasm in first complete remission: An effective therapy for a rare disease. Leuk. Lymphoma. 2016;57:1942–1944. doi: 10.3109/10428194.2015.1115032.
- Gruson B., Vaida I., Merlusca L., Charbonnier A., Parcelier A., Damaj G., Royer B., Marolleau J.-P. l-asparaginase with methotrexate and dexamethasone is an effective treatment combination in blastic plasmacytoid dendritic cell neoplasm. Br. J. Haematol. 2013;163:543–545. doi: 10.1111/bjh.12523.
- Gilis L., Lebras L., Bouafia-Sauvy F., Espinouse D., Felman P., Berger F., Salles G., Coiffier B., Michallet A.-S. Sequential combination of high dose methotrexate and l-asparaginase followed by allogeneic transplant: A first-line strategy for CD4+/CD56+ hematodermic neoplasm. Leuk. Lymphoma. 2012;53:1633–1637. doi: 10.3109/10428194.2012.656627.
- Reimer P., Rüdiger T., Kraemer D., Kunzmann V., Weissinger F., Zettl A., Konrad Müller-Hermelink H., Wilhelm M. What is CD4+CD56+ malignancy and how should it be treated? Bone Marrow Transplant. 2003;32:637–646. doi: 10.1038/sj.bmt.1704215. [PMC free article]
- Kharfan-Dabaja M.A., Lazarus H.M., Nishihori T., Mahfouz R.A., Hamadani M. Diagnostic and therapeutic advances in blastic plasmacytoid dendritic cell neoplasm: A focus on hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2013;19:1006–1012. doi: 10.1016/j.bbmt.2013.01.027.
- Roos-Weil D., Dietrich S., Boumendil A., Polge E., Bron D., Carreras E., Iriondo Atienza A., Arcese W., Beelen D.W., Cornelissen J.J., et al. Stem cell transplantation can provide durable disease control in blastic plasmacytoid dendritic cell neoplasm: A retrospective study from the European Group for Blood and Marrow Transplantation. Blood. 2013;121:440–446. doi: 10.1182/blood-2012-08-448613. [PubMed]
- Aoki T., Suzuki R., Kuwatsuka Y., Kako S., Fujimoto K., Taguchi J., Kondo T., Ohata K., Ito T., Kamoda Y., et al. Long-term survival following autologous and allogeneic stem cell transplantation for blastic plasmacytoid dendritic cell neoplasm. Blood. 2015;125:3559–3562. doi: 10.1182/blood-2015-01-621268.
- Arranto C., Tzankov A., Halter J. Blastic plasmacytoid dendritic cell neoplasm with transient response to pralatrexate. Ann. Hematol. 2017;96:681–682. doi: 10.1007/s00277-016-2907-4.
- Leitenberger J.J., Berthelot C.N., Polder K.D., Pro B., McLaughlin P., Jones D., Duvic M. CD4+ CD56+ hematodermic/plasmacytoid dendritic cell tumor with response to pralatrexate. J. Am. Acad. Dermatol. 2008;58:480–484. doi: 10.1016/j.jaad.2007.12.012.
- Bétrian S., Guenounou S., Luquet I., Demur C., Huynh A., Ysebaert L., Recher C., Huguet F. Bendamustine for relapsed blastic plasmacytoid dendritic cell leukaemia. Hematol. Oncol. 2017;35:252–255. doi: 10.1002/hon.2252.
- Ulrickson M.L., Puri A., Lindstrom S., Cassaday R.D., De Padova N., Becker P.S. Gemcitabine and docetaxel as a novel treatment regimen for blastic plasmacytoid dendritic cell neoplasm. Am. J. Hematol. 2017;92:E75–E77. doi: 10.1002/ajh.24696.
- Khwaja R., Daly A., Wong M., Mahé E., Cerquozzi S., Owen C. Azacitidine in the treatment of blastic plasmacytoid dendritic cell neoplasm: A report of 3 cases. Leuk. Lymphoma. 2016;57:2720–2722. doi: 10.3109/10428194.2016.1160084.
- Laribi K., Denizon N., Ghnaya H., Atlassi M., Besançon A., Pineau-Vincent F., Gaulard P., Petrella T. Blastic plasmacytoid dendritic cell neoplasm: The first report of two cases treated by 5-azacytidine. Eur. J. Haematol. 2014;93:81–85. doi: 10.1111/ejh.12294.
- Frankel A.E., Woo J.H., Ahn C., Pemmaraju N., Medeiros B.C., Carraway H.E., Frankfurt O., Forman S.J., Yang X.A., Konopleva M., et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood. 2014;124:385–392. doi: 10.1182/blood-2014-04-566737. [PMC free article]
- Pemmaraju N., Sweet K.L., Lane A.A., Stein A.S., Vasu S., Blum W., Rizzieri D.A., Wang E.S., Duvic M., Aung P., et al. Results of Pivotal Phase 2 Trial of SL-401 in Patients with Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) Blood. 2017;130:1298.
- Kerr D., Zhang L., Sokol L. Blastic Plasmacytoid Dendritic Cell Neoplasm. Curr. Treat. Options Oncol. 2019;20:9. doi: 10.1007/s11864-019-0605-x.
- Budde L., Song J.Y., Kim Y., Blanchard S., Wagner J., Stein A.S., Weng L., Real M.D., Hernandez R., Marcucci E., et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood. 2017;130:811.
- DiNardo C.D., Pratz K., Pullarkat V., Jonas B.A., Arellano M., Becker P.S., Frankfurt O., Konopleva M., Wei A.H., Kantarjian H.M., et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. doi: 10.1182/blood-2018-08-868752. [PMC free article]
- DiNardo C.D., Rausch C.R., Benton C., Kadia T., Jain N., Pemmaraju N., Daver N., Covert W., Marx K.R., Mace M., et al. Clinical experience with the BCL2-inhibitor venetoclax in combination therapy for relapsed and refractory acute myeloid leukemia and related myeloid malignancies. Am. J. Hematol. 2018;93:401–407. doi: 10.1002/ajh.25000.
- Grushchak S., Joy C., Gray A., Opel D., Speiser J., Reserva J., Tung R., Smith S.E. Novel treatment of blastic plasmacytoid dendritic cell neoplasm: A case report. Medicine (Baltimore) 2017;96:e9452. doi: 10.1097/MD.0000000000009452. [PMC free article]
- Agha M.E., Monaghan S.A., Swerdlow S.H. Venetoclax in a Patient with a Blastic Plasmacytoid Dendritic-Cell Neoplasm. N. Engl. J. Med. 2018;379:1479–1481. doi: 10.1056/NEJMc1808354.