Exercise perturbs homeostasis, alters the levels of circulating mediators and hormones, and increases the demand by skeletal muscles and other vital organs for energy substrates. Exercise also affects bone and mineral metabolism, particularly calcium and phosphate, both of which are essential for muscle contraction, neuromuscular signaling, biosynthesis of adenosine triphosphate (ATP), and other energy substrates. Parathyroid hormone (PTH) is involved in the regulation of calcium and phosphate homeostasis. Understanding the e ects of exercise on PTH secretion is fundamental for appreciating how the body adapts to exercise.
- parathyroid hormone
- calcium metabolism
- phosphorous metabolism
- resistance exercise
- endurance exercise
- training
- skeletal muscle function
- bone metabolism
Exercise perturbs homeostasis, alters the levels of circulating mediators and hormones, and increases the demand by skeletal muscles and other vital organs for energy substrates. Exercise also affects bone and mineral metabolism, particularly calcium and phosphate, both of which are essential for muscle contraction, neuromuscular signaling, biosynthesis of adenosine triphosphate (ATP), and other energy substrates. Parathyroid hormone (PTH) is involved in the regulation of calcium and phosphate homeostasis. Understanding the effects of exercise on PTH secretion is fundamental for appreciating how the body adapts to exercise. Altered PTH metabolism underlies hyperparathyroidism and hypoparathyroidism, the complications of which affect the organs involved in calcium and phosphorous metabolism (bone and kidney) and other body systems as well. Exercise affects PTH expression and secretion by altering the circulating levels of calcium and phosphate. In turn, PTH responds directly to exercise and exercise-induced myokines.
- Introduction
Exercise perturbs homeostasis. Acting as a stressor, the more intense and the longer the exercise bout, the stronger the perturbation. Acute physical activity induces a hormone response that re-establishes homeostasis. The endocrine response to acute exercise occurs over multiple phases and its magnitude depends on the work volume and intensity. In contrast, chronic exercise (training) leads to an adaptation to this stimulus and attenuates the body’s response to acute exercise intensity and volume, without abolishing it, however. At an intensity and volume above an individual’s ability to compensate, maladaptation can lead to an abnormal hormone response and functional failure, which can manifest in overreaching and overtraining syndromes [1].
Calcium-phosphate homeostasis is fundamental for the functioning of all cell types in the body, which is why the levels of calcium and phosphorus in the blood are tightly controlled within a very narrow range. Calcium and phosphorous are also essential for the functioning of striated skeletal muscle and cardiac muscle cells, as well as for neuronal and neuromuscular activity. The homeostatic control of the two elements is important for physical activity and exercise performance. The homeostasis of calcium and phosphorous is maintained by the regulation of the entrance gates (the intestines), the exit gates (the kidneys), and the storehouse (the skeleton). The regulation signal is generated by parathyroid hormone (PTH) and other hormones [2].
PTH is primarily expressed by four small glands located behind the thyroid gland, the parathyroid glands, which are devoted to the control of calcium homeostasis [3]. Despite its importance in physiology and its known role in diverse primary and secondary diseases (i.e., hyperparathyroidism and hypoparathyroidism), PTH has been far less studied in exercise endocrinology than calcium-phosphate metabolism.
- Physical Activity, Exercise, and Training: Terminology and Explanation
In popular parlance, the terms and concepts proper to exercise physiology are commonly misused. A clear explanation of the biological mechanisms involved in exercise-induced homeostatic perturbation and the body’s response is therefore warranted. The terms physical activity, exercise, and training are often interchangeably and sometimes incorrectly used. Physical activity broadly refers to any bodily movement that is produced by skeletal muscles and that results in energy consumption. It is intrinsically associated with activities of daily living; except for involuntary actions, the amount of physical activity an individual performs is subjective. Exercise refers to physical activity that is planned, structured, and repetitive. The two terms are often used synonymously. However, exercise is distinguished by the aim to maintain or improve physical fitness [4]. A single bout of exercise is defined as acute exercise, independent of an individual’s physically active status (e.g., an isolated exercise bout in a sedentary subject vs. an exercise bout within a long-term exercise program in a physically active subject), while exercise bouts continued over time constitute training (exercise training).
Muscle action can be distinguished as static (isometric) and dynamic (isotonic). During isometric action, the muscle generates force but its length remains unchanged: external resistance, the weight of a held object for example, exceeds the force generated by the muscle. Additionally, since no movement occurs, no work is performed despite the expenditure of energy. Work is performed during isotonic action: in concentric contraction, the muscle generates a force that overcomes external resistance and causes the muscle fibers to shorten; in eccentric contraction, the sarcomere shortens and the muscle lengthens due to the opposite movement of the external resistance [5].
Categorized by type of metabolism, exercise and training can be defined as: (1) resistance (or strength needed for weight lifting, discus, or hammer or javelin throw) or short-term power or explosive activity dependent mainly on anaerobic metabolism; and (2) endurance, mid-to-long-term activity dependent mainly on aerobic metabolism (e.g., distance running, road cycling, swimming, and triathlon) [6].
Table 1 presents the characteristics of physical activity and exercise.
Table 1. Characteristics of physical activity and exercise.
|
Voluntary Activity |
|
|
Physical Activity |
Body movement generated by skeletal muscles Variable energy expenditure Positive correlation with physical fitness Life-sustained |
|
Exercise |
Body movement generated by skeletal muscles Variable energy expenditure Positive correlation with physical fitness Planned, structured, repetitive Aimed at maintaining/improving physical fitness |
|
Muscle activity |
|
|
Isometric |
The force generated by the muscle is lower than the external resistance (muscle length unchanged) |
|
Isotonic |
The force generated by the muscle exceeds the external resistance - Concentric: the muscle shortens - Eccentric: the muscle stretches while the sarcomeres shorten |
|
Metabolic activity |
|
|
Resistance |
Short-term power/explosive activity Dependent on anaerobic metabolism |
|
Endurance |
Mid-to-long-term activity dependent mainly on aerobic metabolism |
- Physiology of PTH and Calcium-Phosphate Metabolism
3.1. Synthesis, Secretion, Metabolism, and Mechanisms of Action of the Parathyroid Hormone
PTH is expressed and secreted by the chief cells and the oxyphil cells of the parathyroid glands and regulated by a gene on the short arm of chromosome 11 [7]. It is expressed as 115-amino-acid pre-pro-PTH, which is then cleaved in the Golgi apparatus to yield the mature 84-amino-acid PTH and then stored in granules [8]. PTH mRNA transcription and stability are negatively regulated by extracellular calcium concentrations ([Ca2+]e) by binding to the calcium-sensing receptor (CASR), which displays half-maximal inhibition at about 1 mmol/L [Ca2+]e [9,10]. Moreover, the rate of change in [Ca2+]e drives the robustness of the response (i.e., the more rapid the drop in [Ca2+]e, the greater the magnitude of PTH secretion) [8]. In normocalcemic conditions, only 20% of PTH circulates in its intact form (full-length sequence comprises of aa 1–84, thereafter indicated as PTH(1–84)), while 80% is present as inactive fragments [11]. Increased [Ca2+]e stimulates degradation of PTH within the parathyroid cell, which generates biologically inactive C-terminal fragments (e.g., comprising of the aa sequence from 34 to 84 (PTH(34–84)) or from 37 to 84 (PTH(37–84))) [12]. PTH is also degraded by the liver and cleared by the kidney [13]. Though full-length PTH is the prevalent bioactive circulating form, all PTH-derived peptides retaining the N-terminal have long been recognized as biologically active [14]. Low [Ca2+]e stimulates parathyroid cell proliferation and total secretory capacity, while hypercalcemia may induce parathyroid gland hypotrophy, though it is less associated with hyperplasia [15]. PTH secretion exhibits seasonal and circadian fluctuations synchronous with changes in serum calcium, phosphate, and bone turnover. In addition, an ultradian rhythm exists that comprises of seven pulses per hour, accounts for 30% of basal PTH release, and is highly sensitive to changes in ionized calcium. Acute hypocalcemia induces a selective, several-fold increase in pulse frequency and amplitude, whereas hypercalcemia suppresses the pulsatile secretion component [16].
PTH gene transcription is inhibited by 1α,25-dihydroxyvitamin D (1α,25-(OH)2D), the active form of vitamin D, which also inhibits parathyroid cell proliferation [15,17]. Phosphate, which is closely interrelated with Ca2+ metabolism, plays a role in modulating PTH gene transcription and protein secretion. The endocrine regulation of phosphate is carried out by PTH itself, 1α,25-(OH)2D, and recently discovered bone-derived phosphatonin, fibroblast growth factor (FGF)23 [18]. Mainly secreted by osteocytes, FGF23 plays a central role. Increases in circulating phosphate over days, but not acute increases over hours, upregulates FGF23 secretion, resulting in a phosphaturic effect. Simultaneously, renal 1α,25-(OH)2D production falls, resulting in decreased gut absorption of phosphate and calcium. The overall effect is to prevent hyperphosphatemia and ectopic mineralization. Diurnal variation in serum phosphate is not associated with increases in serum FGF23. This contrasts with diurnal variation in serum Ca2+, which is inversely correlated with changes in serum PTH levels. FGF23 regulates phosphate metabolism, whereas PTH and 1α,25-(OH)2D regulate both phosphate and Ca2+ metabolism. Circulating phosphate concentrations do not directly affect PTH secretion, although high dietary phosphorus intake and oral phosphate supplements do so indirectly by reducing the [Ca2+]o concentration. The three hormones (PTH, 1α,25-(OH)2D, and FGF23) modulate one another’s secretion: FGF23 decreases PTH secretion, 1α,25-(OH)2D decreases PTH secretion and increases FGF23 secretion, and PTH increases FGF23 secretion [19].
Finally, PTH synthesis and secretion are modulated by the transforming growth factor (TGF)-α, prostaglandins (PGs), and cations such as lithium. Figure 1 illustrates the regulation of PTH expression and secretion.
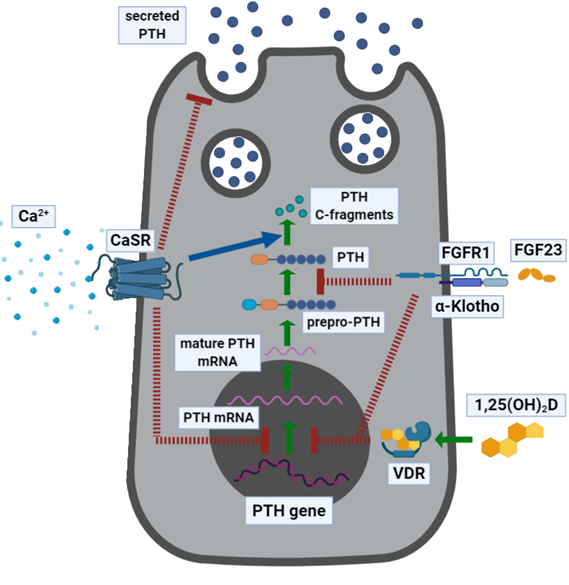
Figure 1. Parathyroid hormone expression and secretion in parathyroid cells. Parathyroid hormone (PTH) is expressed as a prepro-PTH that is then cleaved into mature PTH and stored in granules until secreted. PTH mRNA transcription and PTH secretion are inhibited following the activation of the calcium-sensing receptor (CASR) by extracellular calcium [Ca2+]e, which also stimulates the intracellular inactivation of PTH, into biologically inactive C-terminal fragments, operated throughout the cleavage. Osteocyte-derived fibroblast growth factor (FGF)-23 activates the FGF receptor (FGFR1), heterodimerized with its coreceptor α-Klotho, and inhibits PTH mRNA transcription and PTH protein maturation from prepro-PTH. Finally, 1α,25-dihydroxyvitamin D (1α,25-(OH)2D) binds the intracellular vitamin D receptor (VDR) and inhibits the expression of PTH mRNA. The green arrows indicate PTH expression, the blue arrow indicates a stimulatory pathway, and the red dashed lines indicate inhibitory pathways.
PTH binds through its N-terminal domain a specific class II G-protein coupled receptor (GPCR) expressed by the target tissues, the PTH/PTHrP (PTH related peptide) receptor type 1 (PTHR1) [20,21]. PTHR1 is also targeted by PTHrP, a peptide sharing homology with the N-terminal portion of PTH. Unlike PTH, PTHrP explicates its action in an autocrine/paracrine fashion. Based on PTHR1 expression patterns, the main PTH targets are the bone and the kidney [22]. PTHR1 is coupled with different G-protein classes, resulting in the activation of several intracellular pathways. Gαs activates adenylate cyclase (AC), which results in the synthesis of cyclic AMP (cAMP) and the activation of protein kinase (PK) A. Activated PKA phosphorylates transcription factors, including the cAMP-response element-binding (CREB) protein. CREB phosphorylation regulates its interaction with the cAMP-responsive element (CRE) on DNA and the transcription of target genes. Gαq activates phospholipase (PL) C, which cuts a membrane-enriched phospholipid, phosphatidylinositol-(4,5)-bisphosphate (PIP2), into diacylglycerol (DAG) and inositol-(1,4,5)-trisphosphate (IP3). By binding its receptor in the endoplasmic reticulum, IP3 activates the receptor-gated Ca2+ channel and the release of calcium into the cytoplasm from the endoplasmic reticulum stores. The derived Ca2+ spike allows the translocation of PKC to the plasma membrane, where DAG, released by PLC, activates PKC. Gα12/Gα13 activates PLD [18]. Figure 2 illustrates the PTH-induced signaling pathways.
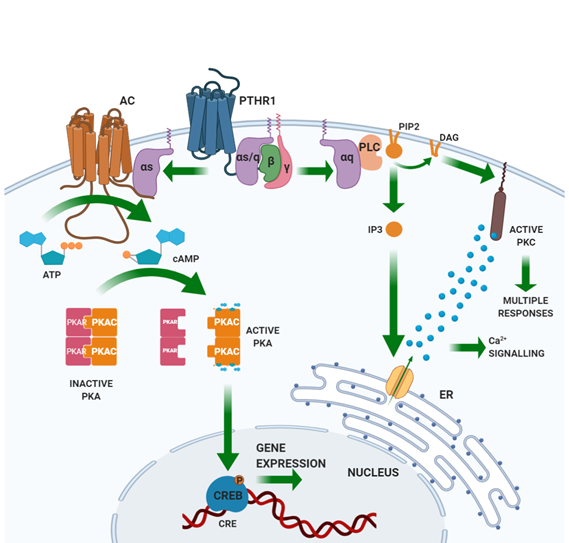
Figure 2. Signaling pathways induced by the activation of the parathyroid hormone receptor. Parathyroid hormone receptor (PTHR)1 is a class II G protein-coupled receptor. Binding of PTH to PTHR1 activates a Gαs protein that activates the adenylate cyclase (AC). AC catalyzes the formation of cyclic adenosine monophosphate (cAMP) from adenosine triphosphate (ATP). cAMP binds and activates protein kinase A (PKA), which, in turn, phosphorylates the cAMP-responsive element binding protein (CREB) into the nucleus, enabling its binding to the cAMP-responsive element (CRE) on DNA and, thus, activates transcription of specific genes. Through the activation of Gαq, PTHR1 activates membrane-associated phospholipase C (PLC), which cleaves the membrane phospholipid phosphatidylinositol-(4,5)-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol-(1,4,5)-trisphosphate (IP3). IP3 diffuses in the cytoplasm, reaches the endoplasmic reticulum, and induces the release of Ca2+ by activating receptor-gated Ca2+ channels. The Ca2+ released from the endoplasmic reticulum activates Ca2+-dependent responses and, together with the DAG produced by PLC, activates protein kinase C (PKC), which mediates intracellular responses.
PTH binding to its cognate receptor induces desensitization, which is initiated by phosphorylation of a serine residue at the C-terminus of PTHR1 mediated by GPCR kinase (GRK). This reaction culminates in the sequestration of the receptor within clathrin-coated endocytic vesicles, where the receptor is still able to activate Gs-mediated signaling and its degradation or recycling to the membrane [23,24].
Finally, the intracellular tail of PTHR1 interacts with adaptor proteins, such as those belonging to the Na/H exchanger regulatory protein factor group (NHERFs) that protect the receptor from downregulation, stimulates its interaction with Gq (stimulation of the PLC-PKC pathway) and inhibits its interaction with Gs (inhibition of the AC-PKA-CREB pathway). Modulation by the NHERFs and activation of the downstream pathway depend upon their expression profile in tissues: in osteoblasts and kidney tubule cells, both Gs- and Gq-dependent pathways can be activated [25–29].
3.2. Parathyroid Hormone and Calcium-Phosphate Homeostasis
The action of PTH is devoted mainly to maintaining [Ca2+]e within the normal range. A [Ca2+]e decrease induces the secretion of PTH from the parathyroid glands. In response, reabsorption of Ca2+ (and Mg2+) in the kidney tubule is increased, while Pi (and HCO3-) reabsorption is inhibited. PTH and hypocalcemia stimulate the AC-PKA-cytochrome P27B1 (CYP27B1) pathway-mediated hydroxylation of 25-hydroxyvitamin D (25-(OH)D) into the active form (1α,25-(OH)2D). In the small intestines, 1α,25-(OH)2D increases Ca2+ (and also Pi) absorption. In the bone, PTH rapidly enhances the turnover rate, thus mobilizing Ca2+ and Pi. Once [Ca2+]e is restored, PTH expression and secretion and activation of vitamin D are again inhibited [30].
3.2.1. The Kidney
Calcium reabsorption in the kidney takes place along the whole nephron, but the major part is taken up in the proximal tubule where most of the solutes and water are resorbed [31]. However, the effects of PTH on calcium reabsorption are limited to the distal portion of the nephron. In the cortical, thick ascending Henle’s loop, 20% of filtered Ca2+ is reabsorbed through activation of PTHR1, which increases the activity of the Na/k/Cl cotransporter, allowing reabsorption of NaCl and, in turn, the paracellular reabsorption of Ca2+ (and Mg2+). In contrast, when [Ca2+]e rises, paracellular calcium resorption is inhibited by the activation of CASR [32–34]: 15% of the filtered calcium is reabsorbed in the distal convoluted tubule via a transcellular route mediated by the apically expressed, highly selective Ca2+ channel transient receptor potential cation channel subfamily V member 5 (TRPV5). Once in the tubule cell, the calcium is translocated to the basolateral membrane by carriers (e.g., calbindin-D28k) and is eliminated by the sodium/calcium exchanger (NCX)1. The PTH-dependent activation of PKC controls the expression and the activity of both TRPV5 and NCX1, as CaSR does for TRPV5 [35–37].
Filtered Pi is reabsorbed only in the proximal tubule, where the epithelial cells express two sodium-dependent phosphate transporters (NaPi-IIa and NaPi-IIc) on the apical side [38]. In this section of the nephron, PTHR1 is expressed [39,40] and associated with both the apical membrane, where it is coupled to the NHERF-dependent PLC-PKC signaling pathway [41], and the basolateral membrane, where it is coupled to the AC-PKA pathway [42]. In both pathways, stimulation by PTH results in downregulation of NaPi-IIa/IIc and reabsorption of Pi [43,44]. Most of the cAMP generated by the activation of tubular PTHR1 is eliminated with the urine and it can be considered a reliable marker of the PTH function [45].
3.2.2. Bone
PTHR1 is expressed by the bone cells of the osteoblastic lineage (including osteoprogenitors, lining cells, immature and mature osteoblasts, and osteocytes) as well as by mesenchymal stem cells (MSC). PTH stimulation enhances the cell proliferation rate, commitment towards osteoblastic differentiation, osteoblastic activity, bone matrix deposition, and mineralization. PTH can also stimulate the expression of the tumor-necrosis factor (TNF)α-related ligand of the receptor activator of nuclear factor κB (RANKL), a stimulating factor for osteoclast differentiation and activity that expresses the receptor for RANKL (RANK) and inhibits osteoprotegerin (OPG), a decoy receptor for RANKL. Osteoclast activation causes bone resorption and the release of Ca2+ and Pi from the resorbing bone. PTH stimulates bone turnover, but the direction of this stimulus toward formation (anabolic) or resorption (catabolic) depends on other factors.
The anabolic role of PTH in bone has been demonstrated in mice in which PTH knockout (KO) was noted to reduce trabecular bone mass and the number of metaphyseal osteoblasts in fetal and neonatal life [46] and in adult life when PTH is needed for fracture healing [47]. In humans, the role of PTH in the bone becomes evident when PTH physiology is more or less severely affected: hypoparathyroidism (parathyroid gland hypofunction associated with reduced secretion of PTH), hyperparathyroidism (parathyroid gland hyperfunction associated with inappropriate secretion of PTH), and PTH administration are all associated with dysregulation of bone remodeling.
PTH stimulates cell proliferation through the cAMP-mediated induction of c-fos in stromal cells and osteoblasts and MAPK cascades in osteoblasts [48–50]. cAMP signaling may also induce Runx2 (also known as Cbfa1), an early transcriptional regulator of osteoblast differentiation [51]. Osteoblast differentiation is also driven by the PTH-induced activation of Tmem119 (Smad3-related factor), which stimulates the bone morphogenic protein (BMP)-Runx2 pathway [52]. PTH-induced osteoblastic differentiation implies the exit from the cell cycle dependent on the inhibition of cyclin D1 expression and the induction of cell cycle inhibitors (p27Kip1 and p21Cip1) [53,54], as well as the inhibition of proapoptotic factors (Bad), the induction of antiapoptotic factors (Bcl) [55], and the increased efficiency of DNA repair [56].
PTH also affects the expression of factors interfering with osteoblast differentiation that are produced by other cells. It inhibits the expression of the unloading-stimulated osteocyte-derived osteoblastogenic inhibitor sclerostin (Sost) [57,58]. PTH induces and stimulates the release of insulin-like growth factor (IGF)-1, an inducer of the osteoblastic pool and osteoblast differentiation [59], and the release of FGF2, which has proproliferative and antiapoptotic effects on osteoblasts [60].
Besides the anabolic effects on bone, when [Ca2+]e is decreased, PTH released from the parathyroid glands induces bone resorption or, better, causes an imbalance in bone turnover towards resorption. This effect seems to be limited to cortical bone, though when blood PTH levels are elevated, the trabecular compartment may also be involved. PTH induces the expression of RANKL in osteoblastic cells and downregulates OPG expression, which stimulates osteoclastogenesis and osteoclast resorption [61,62]. Along with calcium and phosphate, bone matrix resorption causes the release of other factors buried in the matrix during its synthesis (e.g., TGF, BMPs, FGFs and IGF), which, in turn, induces osteoblastogenesis and osteoblastic activity [18]. Furthermore, activated osteoclasts directly recruit osteoblast and osteoblast precursors at the resorption pit by secreting factors (e.g., ephrin, sphingosine-1-phosphate (S1P) and BMP6) [63–66]. The PTH-mediated stimulation of bone resorption is one way by which the body increases the osteoblast pool and stimulates osteoblastic activity and bone anabolism [18].
In patients with osteoporosis, PTH (teriparatide and abaloparatide) is administered intermittently; there is an early phase of enhanced bone formation without resorption (anabolic window), followed by a phase of enhanced turnover (formation and resorption) [67,68]. The temporary extension of the anabolic window allows for a net bone accrual and a gain of up to + 20% in trabecular and endocortical bone, while periosteal bone is mainly subject to the modeling effects of prolonged treatment (less than 10% of bone gain) [69–73].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21155388