Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Surgery
Helices are the most common secondary structural elements in many functional proteins and have attracted much interest when examining the formation kinetics from a coil structure. Measurements describing the timescale to a helix from a coiled structure have been substantially improved with the availability of spectrophotometers with a dramatically improved time resolution. Early studies showed that helix–coil transitions occur in the 100-nanosecond range with a model helix near room temperature.
- laser temperature jump
- molecular dynamics
- dimensionality reduction
- kinetics
1. Introduction
Formation of the functional form of a protein from initiation of a primary sequence and propagation through secondary structural elements is one of the most fundamental biochemical processes in biological systems. Understanding the mechanism of protein structure formation from its amino acid sequence [1][2][3][4][5][6][7][8][9][10][11][12][13][14][15] and predicting the three-dimensional structure from a given sequence [16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36] were two critical questions that attracted scientists from a variety of scientific fields. The quest for a solution to these intellectually challenging questions encouraged an interdisciplinary approach. Significant advances with new experimental approaches and the development of sophisticated theory have provided a much more detailed picture involving the complex pathway of structure formation through folding and the ability to predict three-dimensional structure [37][38]. Experimental studies to characterize the kinetics and thermodynamics of folding in the simplest systems have been very important in establishing a baseline for investigating more complex systems [38][39][40][41]. Experiments with dramatically improved time resolution have facilitated the use of fast kinetic measurements to study the relaxation dynamics of secondary structural elements and single-domain proteins in the sub-nanosecond to millisecond timescale [17][42][43][44][45][46]. These fast kinetic measurements have also been critical in providing a benchmark for a reality check of simulation studies. Analysis of all-atom molecular dynamics simulation results to study relaxation dynamics validated at an experimentally measured very short timescale can potentially provide an atomically detailed description of actual measured kinetic data [47][48][49][50]. Atomic-level descriptions strongly influence the microscopic picture of folding dynamics from molecular dynamics simulation results. It is essential to have measured timescales that can be readily compared with simulations. The advancement of theoretical studies with the energy landscape approach to protein folding was another driving force behind the development of fast-folding kinetic measurements [51][52][53][54][55][56]. Results from free energy surface calculations suggested that the barriers to protein folding should be quite small, and for the fastest folding proteins, the barrier may disappear altogether. This barrierless process may produce non-exponential kinetics, suggesting a non-two-state process during folding events. The relaxation dynamics of secondary structural elements, such as helix, hairpin, turn, and the associated timescale, can essentially capture the global structure formation pathway of a protein [44][57][58].
Helices are the most common secondary structural elements in many functional proteins and have attracted much interest when examining the formation kinetics from a coil structure. Measurements describing the timescale to a helix from a coiled structure have been substantially improved with the availability of spectrophotometers with a dramatically improved time resolution. Early studies showed that helix–coil transitions occur in the 100-nanosecond range with a model helix near room temperature. Ultrafast laser temperature jump spectroscopy has been used to measure the relaxation dynamics of helix formation in 5- to 21-residue and longer helical peptides with picosecond to microsecond resolution [42][43][44][45][46]. These newly measured kinetics with much faster time resolution have been able to identify new details in the helix formation pathway, such as non-exponential kinetic processes [50]. One of the more interesting experimental findings has been the detection of a stable helix formation in alanine-based homopolymers in solution at physiological pH [59]. An additional intriguing finding was obtained through measurement of the relaxation dynamics, previously unavailable, of a five-residue helix forming a heteropeptide with ultrafast laser t-jump, providing an important benchmark that can be easily compared with simulation times [45][46].
An extensive explanation of the approaches to modeling the helix–coil transition was described earlier [47]. A description of this biomolecular phenomenon involving statistical models [60] and the transition path was investigated earlier [61]. A true long-term molecular dynamics simulation can provide an atomically detailed description of kinetics events by extracting the rate of transitions and the nature of microscopic pathways and intermediates and can provide additional input to our theoretical understanding. Recent advancements in theory, more sophisticated algorithms, and a long-term trajectory made possible an atomistic description of kinetics and pathways and validation of experimental results, as well as describing new findings for a wide range of molecular processes [62][63][64][65][66][67][68][69][70][71][72][73].
2. Helix–Coil Kinetics
Helix–coil transitions have been studied experimentally and theoretically for nearly 60 years. In the late 1980s, many peptides comprising similar lengths and sequences found in proteins seemed to form stable helices in an aqueous environment. Since then, a large volume of equilibrium experiments and theoretical studies have been devoted to investigating the formation mechanism of a-helices in the steady state. Alanine-based peptides formed the backbone of these studies as they have a higher propensity to form helices. Kinetic studies, primarily with a 21-residue alanine-based peptide with an attached fluorescent probe at the N-terminus (X-(A)5 (A3RA)3-A-NH2), were performed with a nanosecond laser temperature jump apparatus [43]. The result was inclusive with the presence of an unexplained observed kinetic component. This observation prompted further experiments to understand helix–coil transition kinetics with an improved a-helical heteropeptide Ac-WA3H(A3RA)3-A-NH2, where the first turn of helix formation was monitored via quenching of tryptophan (W) fluorescence by a protonated histidine (H) residue four residues away [44]. Laser t-jump in the nanosecond to microsecond timescale was employed to measure structure formation rates and extract insights into the helix–coil transition pathway after initiation of the temperature jump. At the final temperatures after the jump, measured kinetic traces showed distinct non-exponential kinetics with a fast component clearly above the instrument time resolution of six nanoseconds (Figure 1A–C). Comparison of a single and a bi-exponential fit to the measured data showed that the bi-exponential fit (orange) could completely describe the kinetics instead of the single exponential fit (green). Analysis of the log of rates vs. 1/T showed a nonlinear process (Figure 1D), indicating a deviation from a two-state kinetic process. These experimental observations established the groundwork for the possible presence of intermediates along the transition pathway with the presence of faster kinetic components in this α-helical system. They also prompted an important question of whether these species, when transitioning from coil to helix through the formation of intermediates, lie in a homogeneous or heterogeneous state, pointing to the possible existence of multiple pathways along the transition pathways.
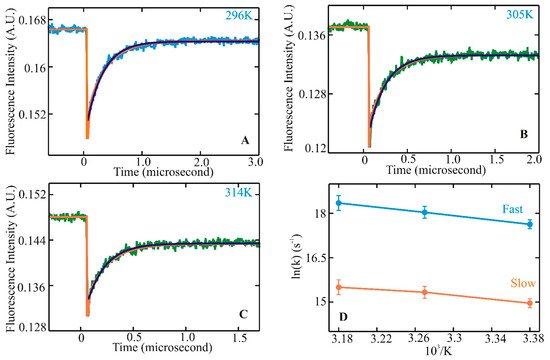
Figure 1. Helix–coil kinetics measured for WH210 following laser t-jumps. Kinetic traces following relaxation dynamics at final temperatures of 296 (A), 305 (B), and 314 K (C). Tryptophan fluorescence intensity change is plotted against the change in time. The single exponential fit to the data is shown in green. The bi-exponential fit to the measured kinetic trace is represented in orange for WH21 at 296, 305, and 314 K. The WH21 concentration in the measured sample is ~100 μM. Kinetic rates in Arrhenius form, ln(k) vs. 1/T, are shown in (D), where the solid blue line represents the fast component, and the slower component is in orange [74].
This entry is adapted from the peer-reviewed paper 10.3390/biom11091351
References
- Bryngelson, J.D.; Wolynes, P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528.
- Bryngelson, J.D.; Wolynes, P.G. Intermediates and barrier crossing in a random energy model (with applications to protein folding). J. Phys. Chem. 1989, 93, 6902–6915.
- Alm, E.; Baker, D. Matching theory and experiment in protein folding. Curr. Opin. Struct. Biol. 1999, 9, 189–196.
- Alm, E.; Baker, D. Prediction of protein-folding mechanisms from free-energy landscapes derived from native structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11305–11310.
- Allew, R.M.; Sabelko, J.; Gruebele, M. Direct observation of fast protein folding: The initial collapse of apomyoglobin. Proc. Natl. Acad. Sci. USA 1996, 93, 5759–5764.
- Ballew, R.M.; Sabelko, J.; Gruebele, M. Observation of distinct nanosecond and microsecond protein folding events. Nat. Struct. Biol. 1996, 3, 923–926.
- Bieri, O.; Wirz, J.; Hellrung, B.; Schutkowski, M.; Drewello, M.; Kiefhaber, T. The speed limit for protein folding measured by triplet-triplet energy transfer. Proc. Natl. Acad. Sci. USA 1999, 96, 9597–9601.
- Blanco, F.J.; Rivas, G.; Serrano, L. A short linear peptide that folds into a β-hairpin in aqueous solution. Nat. Struct. Biol. 1994, 1, 584–590.
- Brooks, C.L. Simulations of protein folding and unfolding. Curr. Opin. Struct. Biol. 1998, 8, 222–226.
- Bryngelson, J.D.; Onuchic, J.N.; Socci, N.D.; Wolynes, P.G. Funnels, pathways, and the energy landscape of protein folding: A synthesis. Proteins: Struct. Funct. Bioinform. 1995, 21, 167–195.
- Buckler, D.R.; Haas, E.; Scheraga, H.A. Analysis of the Structure of Ribonuclease A in Native and Partially Denatured States by Time-Resolved Nonradiative Dynamic Excitation Energy Transfer between Site-Specific Extrinsic Probes. Biochemistry 1995, 34, 15965–15978.
- Burton, R.E.; Huang, G.S.; Daugherty, M.A.; Calderone, T.L.; Oas, T.G. The energy landscape of a fast-folding protein mapped by Ala-->Gly substitutions. Nat. Struct. Biol. 1997, 4, 305–310.
- Burton, R.E.; Huang, G.S.; Daugherty, M.A.; Fullbright, P.W.; Oas, T.G. Microsecond protein folding through a compact transition state. J. Mol. Biol. 1996, 263, 311–322.
- Callender, R.H.; Dyer, R.; Gilmanshin, R.; Woodruff, W.H. FAST EVENTS IN PROTEIN FOLDING: The Time Evolution of Primary Processes. Annu. Rev. Phys. Chem. 1998, 49, 173–202.
- Camacho, J.; Thirumalai, D. Theoretical predictions of folding pathways by using the proximity rule, with applications to bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. USA 1995, 92, 1277–1781.
- Chakrabartty, A.; Baldwin, R.L. Stability of α-helices. Adv. Prot. Chem. 1995, 46, 141–176.
- Chan, C.K.; Hofrichter, J.; Eaton, W.A.; Winkler, J.R.; Gray, H.B. Optical Triggers of Protein Folding. Science 1996, 274, 628–629.
- Chan, C.-K.; Hu, Y.; Takahashi, S.; Rousseau, D.L.; Eaton, W.A.; Hofrichter, J. Submillisecond protein folding kinetics studied by ultrarapid mixing. Proc. Natl. Acad. Sci. USA 1997, 94, 1779–1784.
- Chan, H.S.; Dill, K.A. Protein folding in the landscape perspective: Chevron plots and non-arrhenius kinetics. Proteins 1998, 30, 2–33.
- Chen, E.; Wittung-Stafshede, P.; Kliger, D.S. Far-UV time-resolved circular dichroism detection of electron-transfer-triggered cytochrome c folding. J. Am. Chem. Soc. 1999, 121, 3811–3817.
- Clarke, D.T.; Doig, A.J.; Stapley, B.J.; Jones, G.R. The α-helix folds on the millisecond time scale. Proc. Natl. Acad. Sci. USA 1999, 96, 7232–7237.
- Daggett, V.; Levitt, M. Molecular-dynamics simulation of helix denaturation. J. Mol. Biol. 1992, 223, 1121–1138.
- Daura, X.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Reversible peptide folding in solution by molecular dynamics simulation. J. Mol. Biol. 1998, 280, 925–932.
- Daura, X.; van Gunsteren, W.F.; Mark, A.E. Folding-unfolding thermodynamics of a beta-heptapeptide from equilibrium simulations. Proteins 1999, 34, 269–280.
- Dill, K.A.; Shortle, D. Denatured states of proteins. Annu. Rev. Biochem. 1991, 60, 795–825.
- Dill, K.A.; Stigter, D. Modeling protein stability as heteropolymer collapse. Adv. Prot. Chem. 1995, 46, 59–104.
- Dinner, A.R.; Lazaridis, T.; Karplus, M. Understanding β-hairpin formation. Proc. Natl. Acad. Sci. USA 1999, 96, 9068–9073.
- Dobson, C.M. Protein misfolding, evolution and disease. Trends Biochem. Sci. 1999, 24, 329–332.
- Dobson, C.M.; Sali, A.; Karplus, M. Protein folding: A perspective from theory and experiment. Angew. Chem. Int. Edit. 1998, 37, 868–893.
- Duan, Y.; Kollman, P.A. Pathways to a protein folding intermediate observed in 1-microsecond simulation in aqueous solution. Science 1998, 282, 740–744.
- Dyer, R.B.; Gai, F.; Woodruff, W.H.; Gilmanshin, R.; Callender, R.H. Infrared studies of fast events in protein folding. Acc. Chem. Res. 1998, 31, 709–716.
- Eaton, W.A. Commentary: Searching for “downhill scenarios” in protein folding. Proc. Natl. Acad. Sci. USA 1999, 96, 5897–5899.
- Elove, G.A.; Bhuyan, A.K.; Roder, H. Kinetic mechanism of cytochrome c folding: Involvement of the heme and its ligands. Biochemistry 1994, 33, 6925–6935.
- Fersht, A.R. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; Freeman: San Francisco, CA, USA, 1998.
- Fersht, A.R.; Matouschek, A.; Serrano, L. The folding of an enzyme. I. Theory of protein engineering analysis of stability and pathway of protein folding. J. Mol. Biol. 1992, 224, 771–782.
- Mayor, U.; Guydosh, N.R.; Johnson, C.M.; Grossmann, J.G.; Sato, S.; Jas, G.S.; Freund, S.M.V.; Alonso, D.O.V.; Daggett, V.; Fersht, A.R. The complete folding pathway of a protein from nanoseconds to microseconds. Nature 2003, 421, 863–867.
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589.
- Itzhaki, L.S.; Otzen, D.E.; Fersht, A.R. The structure of the transition state for folding of chymotrypsin inhibitor-2 analyzed by protein engineering methods evidence for a nucleation condensation mechanism for protein folding. J. Mol. Biol. 1995, 254, 260–288.
- Jackson, S.E. How do small singledomain proteins fold? Fold. Des. 1998, 3, R81–R91.
- Jackson, S.E.; Fersht, A.R. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 1991, 30, 10428–10435.
- Williams, S.; Causgrove, T.P.; Gilmanshin, R.; Fang, K.S.; Callender, R.H.; Woodruff, A.W.H.; Dyer, R. Fast Events in Protein Folding: Helix Melting and Formation in a Small Peptide. Biochemistry 1996, 35, 691–697.
- Thompson, P.A.; Eaton, W.A.; Hofrichter, J. Laser temperature jump study of the helix <==> coil kinetics of an alanine peptide interpreted with a “kinetic zipper” model. Biochemistry 1997, 36, 9200–9210.
- Thompson, P.A.; Munoz, V.; Jas, G.S.; Henry, E.R.; Eaton, W.A.; Hofrichter, J. The helix-coil kinetics of a heteropeptide. J. Phys. Chem. B 2000, 104, 378–389.
- Mohammed, O.F.; Jas, G.S.; Lin, M.M.; Zewail, A.H. Primary Peptide Folding Dynamics Observed with Ultrafast Temperature Jump. Angew. Chem. Int. Edit. 2009, 48, 5628–5632.
- Lin, M.M.; Mohammed, O.F.; Jas, G.S.; Zewail, A.H. Speed limit of protein folding evidenced in secondary structure dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 16622–16627.
- Jas, G.S.; Kuczera, K. Computer simulations of helix folding in homo- and heteropeptides. Mol. Simulat. 2012, 38, 682–694.
- Kuczera, K.; Jas, G.S.; Elber, R. Kinetics of Helix Unfolding: Molecular Dynamics Simulations with Milestoning. J. Phys. Chem. A 2009, 113, 7461–7473.
- Kreuzer, S.M.; Elber, R.; Moon, T.J. Early Events in Helix Unfolding under External Forces: A Milestoning Analysis. J. Phys. Chem. B 2012, 116, 8662–8691.
- Jas, G.S.; Middaugh, C.R.; Kuczera, K. Non-Exponential Kinetics and a Complete Folding Pathway of an α-Helical Heteropeptide: Direct Observation and Comprehensive Molecular Dynamics. J. Phys. Chem. B 2014, 118, 639–647.
- Onuchic, J.; Luthey-Schulten, A.; Wolynes, P.G. Theory of protein folding: The energy landscape perspective. Annu. Rev.Phys. Chem. 1997, 48, 545–600.
- Onuchic, J.; Socci, N.D.; Luthey-Schulten, Z.; Wolynes, P.G. Protein folding funnels: The nature of the transition state ensemble. Fold. Des. 1996, 1, 441–450.
- Pande, V.S.; Grosberg, A.Y.; Tanaka, T.; Rokhsar, D.S. Pathways for protein folding: Is a new view needed? Curr. Opin. Struct. Biol. 1998, 8, 68–79.
- Pande, V.S.; Rokhsar, D.S. Is the molten globule a third phase of proteins? Proc. Natl. Acad. Sci. USA 1998, 95, 1490–1494.
- Pande, V.S.; Rokhsar, D.S. Molecular dynamics simulations of unfolding and refolding of a β-hairpin fragment of protein G. Proc. Natl. Acad. Sci. USA 1999, 96, 9062–9067.
- Portman, J.J.; Takada, S.; Wolynes, P.G. Variational theory for site resolved protein folding free energy surfaces. Phys. Rev. Lett. 1998, 81, 5237–5240.
- Muñoz, V.; Thompson, P.; Hofrichter, J.; Eaton, W.A. Folding dynamics and mechanism of β-hairpin formation. Nature 1997, 390, 196–199.
- Jas, G.S.; Eaton, W.A.; Hofrichter, J. Effect of viscosity on the kinetics of α-helix and β-hairpin formation. J. Phys. Chem. B 2001, 105, 261–272.
- Hegefeld, W.A.; Chen, S.E.; DeLeon, K.Y.; Kuczera, K.; Jas, G.S. Helix Formation in a Pentapeptide Experiment and Force-field Dependent Dynamics. J. Phys. Chem. A 2010, 114, 12391–12402.
- Doig, A.J. The alpha-helix as the simplest protein model: Helix-Coil Theory, Stability and Design. In Protein Folding, Misfolding and Aggregation: Classical Themes and Novel Approaches; Muñoz, V., Ed.; Royal Society of Chemistry: London, UK, 2008.
- Huo, S.; Straub, J.E. Direct Computation of Long Time Processes in Peptides and Proteins: Reaction Path Study of the Coil-to-Helix Transition in Polyalanine. Proteins 1999, 36, 249–261.
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: Oxford, UK, 1987.
- Allen, R.J.; Frenkel, D.; ten Wolde, P.R. Forward flux sampling-type schemes for simulating rare events: Efficiency analysis. J. Chem. Phys. 2006, 124, 194111.
- Aristoff, D.; Bello-Rivas, J.M.; Elber, R. A mathematical framework for exact milestoning. Multiscale Model. Simul. 2016, 14, 301–322.
- Bahar, I.; Lezon, T.R.; Yang, L.W.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42.
- Bello-Rivas, J.M.; Elber, R. Exact milestoning. J. Chem. Phys. 2015, 142, 94102.
- Bello-Rivas, J.M.; Elber, R. Simulations of thermodynamics and kinetics on rough energy landscapes with milestoning. J. Comput. Chem. 2016, 37, 602–613.
- Bolhuis, P.G.; Chandler, D.; Dellago, C.; Geissler, P.L. Transition path sampling: Throwing ropes over rough mountain passes, in the dark. Annu. Rev. Phys. Chem. 2002, 53, 291–318.
- Bowman, G.R.; Pande, V.S. An Introduction to Markov State Models and Their Applications to Long Timescale Molecular Simulations; Springer: Berlin, Germany, 2014.
- Cardenas, A.E.; Elber, R. Computational study of peptide permeation through membrane: Searching for hidden slow variables. Mol. Phys. 2013, 111, 3565–3578.
- Cardenas, A.E.; Jas, G.S.; DeLeon, K.Y.; Hegefeld, W.A.; Kuczera, K.; Elber, R. Unassisted transport of N-acetyl-L-tryptophanamide through membrane: Experiment and simulation of kinetics. J. Phys. Chem. B 2012, 116, 2739–2750.
- Cardenas, A.E.; Elber, R. Modeling kinetics and equilibrium of membranes with fields: Milestoning analysis and implication to permeation. J. Chem. Phys. 2014, 141, 54101.
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. A Computational Mi-croscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452.
- Jas, G.S.; Kuczera, K. Helix-Coil Transition Courses through Multiple Pathways and Intermediates: Fast Kinetic Measurements and Dimensionality Reduction. J. Phys. Chem. B 2018, 122, 10806–10816.
This entry is offline, you can click here to edit this entry!