Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Genetics & Heredity
Theoretically, a DNA sequence-specific recognition protein that can distinguish a DNA sequence equal to or more than 16 bp could be unique to mammalian genomes. Long-sequence-specific nucleases, such as naturally occurring Homing Endonucleases and artificially engineered ZFN, TALEN, and Cas9-sgRNA, have been developed and widely applied in genome editing.
- CRISPR
- Cas9
- genome/gene-editing
- extracellular vesicles
1. Preface
Besides the common gene knockout purpose, a major goal of genome/gene editing experiments is to precisely convert a selected DNA sequence into a new desired one in the native context of the whole genome. Before the advent of programmable nucleases, most of the cases of the site-specific changes by homology-directed repair (HDR) were executed under the single agent gene editing strategy utilizing single-stranded DNA oligonucleotide templates [1]. The efficiency of HDR could be highly improved when DNA cleavage occurred at or near the recombination site [2,3]. As a comparison of the ranges of genome sizes, those for amphibians and flowering plants are from 1E9 to 1E11 and from 1E8 to 1E11 bp, those of birds and mammals are quite narrow, being around 1E9 and 3E9 bp per ploidy, respectively [4,5]. If we want to precisely operate the genomes of livestock, such as cows, pigs, sheep, and chickens, a nuclease which is able to recognize DNA sequences of equal to or more than 16 bp, theoretically, is indispensable. (let 4n ≥ 3E9, then n ≥ 16). Natural or engineered site-specific nucleases suitable for these purposes and their applications on genome/gene editing are described in this paper.
2. Endonucleases for Genome Editing
2.1. Homing Endonuclease and Meganuclease
Since the original study in 1971 to determine the “ω” self-splicing intervening sequence, it was later recognized as a group I intron [6] located within the mitochondrial gene-encoding large ribosomal RNA of yeast, Saccharomyces cerevisiae [7]. Additionally, the inheritance of this intron was induced by a site-specific endonuclease (now termed I-SceI) encoded within the intron sequence [8]. Such nucleases which can recognize a unique DNA target site among the whole genome are now nomenclatured as Homing Endonucleases, defined as microbial DNA-cleaving enzymes that mobilize their own reading frames by generating double-strand breaks at specific genomic invasion sites. These proteins display an economy of size and yet recognize long DNA sequences (typically 20 to 30 base pairs) [9]. Based on the specific amino acid motif in the catalytic core, HEs were categorized into at least six families, with each being associated with a particular host range. HNH, GIY-YIG, and EDxHD families are largely constrained to phage hosts. PD-(D/E)xK, His-Cys Box, and LAGLIDADG families are encoded in bacterial, protistic, and archaeal/eukaryotic hosts, respectively [10,11,12]. The endonucleases of the LAGLIDADG HE family are often referred to as “meganucleases (MN)” [13]. The LAGLIDADG endonucleases exist both as homodimers (where the two identical protein subunits are each typically 160 to 200 residues in size with a αββαββα core fold in which a long anti-parallel pair of β strands are fitted into the major groove of recognition site), and as monomeric proteins, where a tandem repeat of two LAGLIDADG domains is connected by a variable peptide linker. Compared with their homodimeric cousins, the monomeric proteins are rather small; their individual domains are often only 100 to 120 residues in size and can recognize fully asymmetric DNA target sites. In addition to the benefits that the recognition site of MNs are quite long (18–24 bp) and highly specific and the sizes are small so as to be prepared and engineered [14,15] easily, the richest natural sources are another advantage. For the full spectrum of the mammalian genome, a bank of 3 billion MNs is theoretically needed to cover all possible recognition sites, or hundreds of thousands to overlay all genes. That is a tremendous task.
2.2. Zinc Finger Nuclease (ZFN)
In comparison to a typical type II restriction enzyme, whose recognition site is overlapped with a cutting site, in the type IIS, where “S” means shifted cleavage, the enzyme contains independent modules for a separated recognition site and cutting site, e.g., FokI contains a 382 a.a. N-terminal DNA recognition domain and a 196 a.a. C-terminal nuclease domain (FN) [16]. After the proof-of-concept work prepared chimeric restriction endonuclease by linking the site-specific DNA-binding homeodomain of Ubx with FN [17], the primitive concept of combining the site-specific DNA-binding zinc finger domains with FN, termed as ZFN hereafter, to be an editable hybrid restriction enzyme was launched by Dr. Srinivasan Chandrasegaran’s group at Johns Hopkins [18]. The zinc finger is a big superfamily of domains and C2H2 is the most common type of it. A β sheet-turn-β sheet-turn-α helix has a rigid structure, where two cysteine residues located within the first turn and two histidine residues at the C-terminus of the α helix are coordinated to chelate a zinc ion. The α helix is fitted into the major groove of the DNA double helix and the -1, 3, and 6 a.a. residues interact with three consecutive nucleotides of the sense strand in the 3′ to 5′ orientation, respectively [19]. The C2H2 zinc fingers can be recognized as independent trinucleotide binding modules and linked into a polypeptide to distinguish longer DNA binding. In theory, one can design a zinc finger for each of the 64 possible combinations of trinucleotides, and one can arrange such zinc fingers to compose a sequence-specific artificial protein for any segment of DNA [20]. Because the nuclease activity of FN is stringently present in a dimer form [21], a pair of ZFNs with recognition sites in a tail-to-tail orientation was demonstrated necessary to perform effective double-strand cutting activity, which was essential to enhance the probability of homologous recombination a thousand-fold in vivo [22]. The D483 and R487 residues of FokI were involved in the FN dimer formation by interacting with each other between the two subunits. FN carried a D483R mutation which led both of the 483 and 487 residues of FokI to be positively charged, termed as FNRR. On the other hand, FN carried a R487D mutation, which led both the 483 and 487 residues of FokI to be negatively charged, termed as FNDD. Unlike the wild-type FN, FNRR and FNDD cannot form homodimers themselves; however, they can form heterodimers efficiently [23]. Such phenomena were utilized to improve the cutting specificity by using a pair of complementary FNs for the up-stream and down-stream recognition arm, respectively, as well as to reduce the toxicity caused by the off-targeting by unwanted homo-dimer [24]. Although ZFN seems a powerful tool for genome editions, some drawbacks should be noted. Firstly, there are still no perfect matches between zinc finger proteins and DNA triplexes. The specificity between GC-rich triplexes and zinc fingers was calculated to be 73%, whereas it was only 50% for AT-rich triplexes and their zinc finger partners [25]. The DNA-binding specificity of a C2H2 zinc finger was also revealed to be influenced by neighboring ones [26].
2.3. Transcription Activator-Like Effector Nuclease (TALEN)
Since the isolation of the avrBs3 gene in the bacterial plant pathogen Xanthomonas campestris pv. Versicatoria [27], AvrBs3 protein was found to be injected into plant cells via a type III secretion system [28,29] and to act as a site-specific transcription activator-like effector (TALE) to reprogram host cells. The AvrBs3 protein is composed of 1163 a.a. with a translocation domain at the most N-terminal end of a 287 a.a. N-terminal region, a middle part which contains 17 units of 34 a.a. complete repeats following with a 20 a.a. half repeat, and a 278 a.a. C-terminal region containing nuclear localization signals (NLSs) and an acidic transcriptional activation domain (AD) [30]. The sequences of the 34 a.a. repeats performing an α helix-random coil-α helix structure are nearly identical, except the polymorphic 12th and 13th residues, which are known as the repeat variable di-residue (RVD) and specifically specify a single binding site nucleotide through direct interactions [31,32,33,34]. The specificity and affinity for each RVD to a nucleotide were systemically studied [35,36]. Theoretically, merely four kinds of repeat, each with RVDs recognizing G, A, T, and C, respectively, are necessary to construct a DNA-binding domain specific to any given sequence. The N-terminus 152 a.a. and C-terminus 215 a.a. of AvrBs3 protein could be removed to leave a core region with intrinsic DNA-binding activity, and this core region was used as a fundamental framework for TAL effector nuclease (TALEN) designation [37,38,39,40]. X-ray crystal data revealed that the amino acid residues 162 to 288 of AvrBs4 perform four cryptic repeats of helical bundles to interact with a nucleotide T on the sense strand of a DNA double helix immediately in front of the nucleotides recognized by the canonical repeats, and this region provides the majority of the energy required for high-affinity target binding [41].The RVDs of complete repeat modules bind consecutive nucleotides of sense strands in the 5′ to 3′ direction. The first residue in each RVD (the 12th of the repeat) orients away from DNA to interact with the backbone of the eighth residue of the repeat to stabilize the interhelical loop and allow the second residue of the RVD to project into the major groove of the DNA and make sequence-specific contact with a single nucleotide of the sense strand [42]. The most common RVDs are HD, NG, NN, and NI for C, T, G > A, and A, respectively, in which NN can be replaced by NH for NH is more specific to G but has less affinity [34,35,36,43]. Online tools for custom TALEs and TALENs, such as TALE-NT 2.0, designation and modular assembly methods relying on Golden Gate cloning have been developed, enabling researchers to make constructs in a few days [44,45,46,47,48]. Besides the FN nuclease domain, transcription regulatory domains and DNA modification enzymes can be engineered to the C-terminus of the sequence-specific TALE-core structure to create artificial gene regulatory factors [49,50].
2.4. CRISPR/Cas Nucleases
It was identified that the spacer sequences between identical repeats of the clustered regularly interspaced short palindromic repeat (CRISPR) loci of bacterial genomes might originate from plasmid and phage [51,52]. The CRISPR RNA and CRISPR-associated protein (Cas) systems are now confessed as key components governing bacterial adaptive immune response which consists of three main stages: adaptation, expression, and interference. When a bacterium was attacked by an invader, a short DNA fragment, termed a protospacer, which is neighbored by a protospacer-adjacent motif (PAM) of the invader, was processed by adaptation Cas members, such as Cas1 and Cas2, to be inserted into the 5′ end of a spacer-repeat CRISPR array embedded in the host genome as stored memory. Memory was retrieved as the CRISPR array was transcribed to produce a long precursor CRISPR RNA (pre-crRNA), which was processed by an expression factor, such Cas6 or RNase III, within the repeat region to create mature crRNA, which was incorporated with Cas effectors, such as Cas5, Cas7, Cas8, and Cas11, to yield an RNA-guided sequence-specific endonuclease in the interference stage [53,54]. According to the number of Cas protein subunits included in the effector endonuclease complex, the CRISPR-Cas systems belong to two classes, with multi-subunit effector complexes in class 1, which can be further divided into three types: type I, type III and type IV, and single-protein effectors in class 2, including type II, type V and type VI [55,56,57]. Besides crRNA and Cas9 protein, a trans-activating CRISPR RNA (tracrRNA) whose 5′ region is complemented with the repeat sequence of crRNA is critical to perform endonuclease activity in the type II CRISPR systems. The crRNA and tracrRNA could be engineered into one single-guided RNA (sgRNA) in accompaniment with Cas9 to restore full and specific endonuclease activity [58]. The best-characterized and applied Cas9 enzyme was originally isolated from Steptococcus pyrogenes, and was referred to as SpCas9, or even simply as Cas9. SpCas9 is a large 1368 a.a. multidomain protein with two distinct lobes: the recognition (REC) lobe and the nuclease (NUC) lobe, connected through an arginine-rich bridge helix (residue 56 to 93) and a disordered loop (residue 712 to 717). The REC lobe is composed of three α-helical domains (Hel-I, Hel-II, and Hel-III) and the NUC lobe contains HNH and RuvC-like nuclease domains, as well as a PAM-interacting (PI) C-terminal domain [59,60] (Figure 1A,B). The apo-Cas9 protein should be assembled with guide RNA (native crRNA-tracrRNA hybrid or sgRNA) to achieve site-specific DNA recognition and cleavage activities. The 20 nt spacer sequence of crRNA provided DNA target specificity and the tracrRNA conferred a crucial role in Cas9 protein recruitment. Once the PAM (NGG for SpCas9) directly adjacent to a protospacer target site was trapped by R1333 and R1335 of the Cas9-guide RNA complex, it triggered local DNA melting at the PAM-adjacent site. The PAM-proximal 10–12 nucleotides (nt), 3′-end of the 20 nt spacer sequence is absolutely critical for site specificity, and was referred to as seed region. The DNA cleavage activity of CRISPR-Cas9 was excited by the conformational change induced by the R-loop formation between target DNA and spacer RNA [61,62]. The target DNA strand complementary to spacer RNA was cut by the HNH nuclease domain and the non-target DNA strand by the RuvC nuclease domain to produce a blunt-ended double-strand breakage at 3 bp upstream to PAM [63,64]. Either D10A [58] or H983A [65] mutation destroyed the RuvC nuclease activity. On the other hand, D839A [66], H840A [58], and N863A [67] mutations could eliminate the HNH nuclease activity. These mutations did not influence the target site binding affinity of Cas9-sgRNA. Cas9 carrying the D10A mutation and D10A/H840A double mutations were termed nickase (nCas9) and dead enzyme (dCas9), respectively (Table 1). The dCas9 could be taken as a guide RNA-derived sequence-specific DNA-binding protein, like TALE described above, and coupled with DNA manipulation enzymes or transcriptional activating/inhibitory domains to be harnessed for various applications [64]. The amino acid residues interacting with the PAM bases could be engineered to generate new PAM so as to broaden the spectrum of target sites. Based on the structure-guided rational design, the wild-type D1135, R1335, and T1337 were converted to E, Q, and R, respectively; the PAM was shifted from NGG to NGA. Additionally, as D1135, G1218, R1335, and T1337 were converted to V, R, E, and R, respectively; the PAM became NGC [68]. An engineered SpCas9 bearing D1135L/S1136W/G1218Q/E1219Q/R1335Q/T1337R substitutions in PI domain (SpG) targeted NGN PAM. SpG was further engineered to carry A61R/L1111R/N1317R/A1322R/R1333P substitutions to near-PAMless (NRN > NYN) variants, termed SpRY, with full endonuclease activities [69] (Table 1).
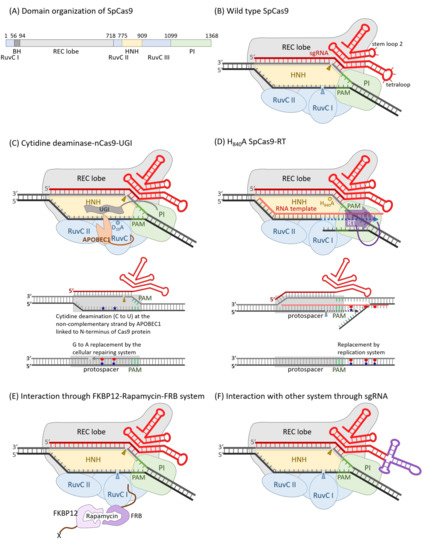
Figure 1. Diagrams of SpCas9 and its derivatives for various applications. The domain organization of SpCas9 (A) and a schematic diagram of wild-type SpCas9 associated with a sgRNA (B) was illustrated. The non-complementary strand is cut by the RuvC nuclease domain, and this nuclease activity was blocked in D10A mutant. On the other hand, the complementary strand was digested by the HNH nuclease domain, and such nuclease activity was destroyed in H840A mutant. (C) The D10A mutant, also named Cas9 nickase (nCas9), was engineered as a C to T nucleotide editor by linking a cytidine deaminase, APOBEC1, on the N-terminus of it and the switching probability could be elevated by the fusion of a uracil glycosylase inhibitor (UGI) on the C-terminus of nCas9. Like TALE, dCas9 could be guided by a sgRNA as a sequence-specific DNA-binding riboprotein. Transcriptional regulators, DNA modification enzymes, or histone modification enzymes could be fused to either or both of the N- and C-termini. In case of reverse transcriptase, it was fused to the C-terminus of Cas9, accompanied by an RNA template with 3′-end complementary to the non-complementary strand of protospacer, which could alter the nearby nucleotides downstream the RuvC cutting site (D). The localization of the Cas9-sgRNA also could be guided to an X-protein through an FKBP12–rapamycin–FRB bridge (E). The localization of the Cas9-sgRNA could also be guided via certain specific interactions, such as those between aptamer RNA and ABP. The tetraloop was replaced by an RNA aptamer of unique secondary structure, which can be recognized by a specific aptamer binding protein (F).
Table 1. The engineered mutations of SpCas9 on target site recognition and nuclease activities.
| Domain Where Mutations Engineered | Functional Changes | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutants | RuvC-I (1–55) |
BH (56–93) |
REC Lobe (94–717) |
RuvC-II (718–774) |
HNH (775–908) |
RuvC-III (909–1098) |
PI (1099–1368) |
||
| SpG | D1135L, S1136W, G1218Q, E1219Q, R1335Q, T1337R |
The recognition sequence of PAM changed from NGG to NGN. | [69] | ||||||
| SpRY | A61R | L1111R, D1135L S1136W, G1218Q E1219Q, N1317R, A1322R, R1333P, R1335Q, T1337R |
The recognition sequence of PAM changed from NGG to PAMless (NRN > NYN). | ||||||
| eSpCas9(1.1) | K848A | K1003A R1060A |
eSpCas9(1.1) displayed efficient and precise genome editing in human cells. | [70] | |||||
| SpCas9-HF1 | N497A R661A Q695A |
Q926A | SpCas9-HF1 performed with high on-target activity and reduced off-target editing. | [71,72] | |||||
| nSpCas9 | D10A | RuvC nuclease activity was eliminated. (nickase) | [58] | ||||||
| SpCas9(H840A) | H840A | HNH nuclease activity was diminished. | |||||||
| dSpCas9 | D10A | H840A | Dead SpCas9 lost both of the RuvC and HNH nuclease activities. | ||||||
| SpCas9(N863A) | N863A | HNH nuclease activity was eliminated. | [67] | ||||||
| SpCas9(D839A) | D839A | HNH nuclease activity was diminished. | [66] | ||||||
| SpCas9(H983A) | H983A | RuvC nuclease activity was eliminated. | [65] | ||||||
Besides Cas9, which recognized G-rich PAM at 3′ end of protospacer, class 2 type V Cas12a (originally called Cpf1) effector enzymes also became attractive [73]. The long pre-crRNA was bound and processed by an intrinsic RNase activity of Cas12a protein to mature crRNA, which was composed of a repeat sequence at the 5′ end and spacer at the 3′ end. This characteristic was utilized to design multiple crRNA in a single RNA transcript [73,74]. A canonical TTTV PAM was at the 5′ end of a 23 bp protospacer. Only a short 42–44 nt crRNA, which was composed of 19 nt repeat and 23–25 nt spacers, was necessary to guide the Cas12a’s RNA-dependent endonuclease activity, of which DNA was cut at the PAM-distal end to leave 5′ protruding staggered ends. Like Cas9, the RuvC nuclease domain was involved in non-complementary strand cleavage, while a new Nuc domain, instead of the HNH domain, was used in Cas12a for complementary strand cleavage [75]. The size of Lachnospiraceae bacterium MA2020 Cas12a (LbCas12a) was merely 1206 a.a. and as active as the most widely used Cas12a isolated from Acidaminococcus sp. (AsCas12a, 1307 a.a.). Engineered LbCas12a with Q571K and C1003Y mutations, referred to as Lb2Cas12a, was more active and could recognize both TTTV and CTTV PAM motives [76].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22189872
This entry is offline, you can click here to edit this entry!