The gastrointestinal tract secretes gut hormones in response to food consumption, and some of these stimulate insulin secretion. Glucagon-like peptide-1 (GLP-1) is an incretin peptide hormone released from the lower digestive tract that stimulates insulin secretion, suppresses glucagon secretion, and decreases hunger. GLP-1 receptor agonist (GLP-1RA) mimics the action of endogenous GLP-1, consequently reversing hyperglycemia and causing weight reduction, demonstrating its efficacy as an antidiabetic and antiobesity agent. Previously restricted to injection only, the invention of the absorption enhancer sodium N-(8-[2-hydroxybenzoyl]amino) caprylate resulted in the development of oral semaglutide, the first ingestible GLP-1RA. Oral semaglutide demonstrated its efficacy in glycemic management and body weight loss with a low risk of hypoglycemia as a monotherapy and in combination with other hypoglycemic medications in its clinical trial programs named Peptide Innovation for Early Diabetes Treatment. Consistent with other injectable GLP-1RAs, gastrointestinal side effects were often reported. Additionally, cardiovascular safety was established by demonstrating that oral semaglutide was not inferior to placebo in terms of cardiovascular outcomes. Thus, oral semaglutide represents a novel treatment option that is particularly well-suited for patients with type 2 diabetes and/or obesity.
1. Introduction
Type 2 diabetes (T2D) is a chronic metabolic condition characterized by beta-cell dysfunction and insulin resistance that worsens over time [
1]. T2D is treated with a combination of lifestyle adjustments, such as diet and exercise, as well as medication intervention [
1]. Over the recent decade, new antidiabetic drugs have been introduced, expanding T2D therapy choices while also increasing treatment complexity [
1,
2]. Along with the development of novel agents, recent guidelines stress the patient’s coexisting diseases and risk factors before glycemic control, recommending medications with cardiovascular and renal benefits over those with glycemic control [
2,
3,
4].
Obesity is a substantial risk factor for having T2D, and the prevalence of T2D is projected to rise as the world’s obese population grows [
5]. Obesity is a modifiable risk factor, hence, correcting the etiology of obesity and insulin resistance may help prevent and treat T2D [
5]. Although lifestyle changes, including diet and exercise, are essential for T2D treatment, most patients with T2D require the use of antidiabetic drugs to achieve their glycemic goals. Metformin, sulfonylureas, meglitinides, thiazolidinediones, alpha-glucosidase inhibitors, and insulin are examples of traditional agents [
1]. While these medications can effectively lower blood glucose levels, many have limitations due to weight gain and hypoglycemia [
1]. Furthermore, only metformin and thiazolidinediones are used to improve insulin sensitivity and affect the pathophysiology of T2D [
1]. As a result, unmet needs in the treatment of T2D persist, necessitating additional research to develop new treatment choices.
1.1. Gut Hormones: The Metabolism Regulators
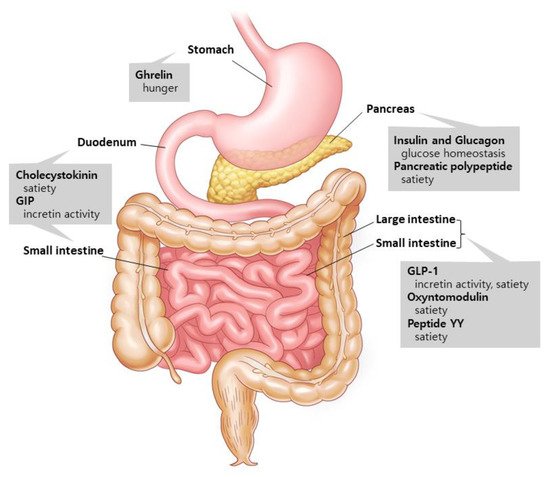
Gut hormones are involved in metabolism and interact with one another to digest the nutrients that are consumed [
6] (
Figure 1). Two major hormones that induce insulin production are glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) [
7]. Ghrelin, on the other hand, is a stomach hormone that inhibits insulin secretion by releasing growth hormone [
8]. Cholecystokinin, GLP-1, and peptide YY are hormones that slow down stomach emptying and suppress appetite [
9,
10]. Glucagon is produced in the pancreatic alpha cells and increases hepatic glucose synthesis and lipolysis to boost blood glucose levels [
11]. Pancreatic polypeptide excreted from the pancreas is involved in the long-term regulation of appetite [
10]. Oxyntomodulin, released concurrently with GLP-1 by the L-cells, reduces food intake while increasing energy consumption and insulin secretion [
12,
13]. The aforementioned gut hormones are being studied in order to find new potential medications to address the unmet demand in T2D treatment.
Figure 1. Gut hormones and their supposed actions. Ghrelin is released by the stomach. Insulin, glucagon, and pancreatic polypeptide are excreted from the pancreas. Cholecystokinin and GIP are secreted in the duodenum and small intestines, and GLP-1, oxyntomodulin, and peptide YY are released by the small and large intestines. These hormones from the gastrointestinal tract communicate with the peripheral and central nervous systems to control a variety of metabolic activities. Modified from Murphy et al. [
6].
1.2. Glucagon-like Peptide-1 (GLP-1): An Innovator for Gut Hormone Therapies
The release of incretin hormones after eating a meal has opened new avenues for the development of antidiabetic drugs [
14]. Incretins promote insulin release from pancreatic beta cells in response to hyperglycemia, preserving normoglycemia [
15]. Both GIP and GLP-1, which are each released by K-cells in the duodenum and upper jejunum and by L-cells in the distal ileum and large intestines, enhance glucose-mediated insulin release [
16,
17]. In patients with T2D, however, GIP loses much of its insulinotropic activity, whereas GLP-1 demonstrates a sustained but diminished insulinotropic response [
18]. Furthermore, while GLP-1 reduces glucagon secretion in a glucose-dependent manner, GIP has no effect on glucagon secretion during hyperglycemia and instead increases it during hypoglycemia [
19]. GLP-1 has also been demonstrated to have pleiotropic effects, such as lowering hunger and food intake, as well as slowing stomach emptying and small bowel movement [
20,
21]. As a result, GLP-1 has become a hot target for possible T2D and obesity medicines.
GLP-1 receptor agonist (GLP-1RA) decreases food intake by increasing gastric emptying time and satiety, resulting in body weight loss [
22]. Despite its antidiabetic and antiobesity properties, the GLP-1RA has been restricted in usage because of gastrointestinal side effects (nausea, vomiting, constipation, and abdominal discomfort) [
7]. Furthermore, despite its established efficacy, the subcutaneous injection technique of administering GLP-1RA has further limited its prescription. Concerns with injection, including pain and fear, have been noted in studies as barriers to maintaining GLP-1RA in real-world practice [
23,
24]. As a result, the first oral GLP-1RA semaglutide is a significant achievement, offering a practical alternative to injectables for patients with T2D.
2. Pharmacodynamics and Pharmacokinetics of Oral Semaglutide
Semaglutide was originally designed as a once-weekly subcutaneous long-acting GLP-1RA. Semaglutide is a human GLP-1 analog with 94% similarity to natural human GLP-1 but has amino acid changes that improve albumin binding, decrease renal clearance, and boost resistance to DPP-4 destruction [
25]. Semaglutide demonstrated efficacy in glycemic control and body weight reduction compared to placebo and active comparators, such as sitagliptin, exenatide extended-release, dulaglutide, and insulin glargine, in the Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) clinical trials [
26,
27,
28,
29,
30]. Furthermore, semaglutide improved cardiovascular outcomes significantly [
31].
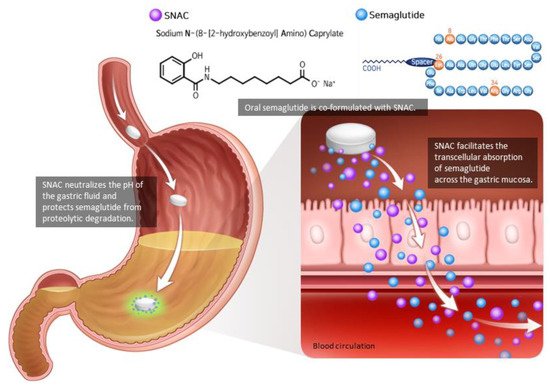
As the peptide-based drug is degraded by proteolytic enzymes and the pH of the gastrointestinal tract, semaglutide must be injected subcutaneously. Semaglutide could be developed as an oral tablet by combining it with an absorption enhancer known as sodium N-(8-[2-hydroxybenzoyl]amino) caprylate (SNAC) [
32,
33] (
Figure 2). In a concentration-dependent manner, SNAC forms a noncovalent bond with GLP-1, increasing lipophilicity and transcellular absorption of semaglutide through the stomach epithelium [
32,
34]. Additionally, in the acidic environment of the stomach, SNAC acts as a local pH buffer for semaglutide, increasing solubility and protecting the drug from degradation [
34]. As SNAC’s activity is brief and reversible, it separates from the medication once it reaches the bloodstream [
32].
Figure 2. Oral semaglutide and SNAC. Oral semaglutide must be co-formulated with the absorption enhancer SNAC in order to be absorbed. SNAC raises the local pH, resulting in increased solubility and protection from proteolytic degradation. SNAC promotes the absorption of semaglutide across the gastric mucosa in a time- and concentration-dependent manner, which is totally reversible. Modified from Andersen et al. [
33].
In contrast to most other medications absorbed in the intestines, oral semaglutide has a distinct pharmacokinetic profile since it is virtually fully absorbed in the stomach. After 15–35 min of oral intake, the medication reaches its maximal concentration [
32]. As food interferes with drug absorption, oral semaglutide should be given while fasting [
35]. Semaglutide exposure was unaffected by the amount of water used to take the drug [
35]. However, the higher the semaglutide exposure, the longer the post-dose fasting time; hence, at least 30 min of post-dose fasting is recommended [
35]. Renal and hepatic impairment had no effect on the pharmacokinetics of oral semaglutide, indicating that individuals with renal and hepatic impairment do not require dose adjustments [
36,
37]. In investigations involving omeprazole, lisinopril, warfarin, digoxin, metformin, levonorgestrel, ethinyl estradiol, furosemide, and rosuvastatin, no significant drug–drug interactions were found [
38,
39,
40]. Thyroid function tests should be monitored in patients receiving both oral semaglutide and levothyroxine since the pharmacokinetics of levothyroxine are influenced by a 33% increase in exposure when taken with oral semaglutide [
41].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22189936