Endogenous retroviruses (ERVs) are long terminal repeat (LTR)-retroelements of the Retroviridae genus. ERVs reside in vertebrate genomes, are particularly abundant in mammals, and still actively retrotranspose in mice. This entry describes their relationship to other LTR-retroelements and how they replicate. A brief introduction into epigenetic reprogramming of ERVs is followed by a few examples of how reprogramming of ERVs assists embryonic development in mouse and human.
- endogenous retrovirus (ERV)
- long terminal repeat (LTR)
- retrotransposon
- tRNA
- epigenetic reprogramming
1. Definition
Endogenous retroviruses (ERVs) are one type of long terminal repeat (LTR)-retroelements. LTR-retroelements include Pseudoviridae (Ty1, Copia), Metaviridae (Ty3/Gypsy), and Retroviridae such as ERVs, human immunodeficiency virus (HIV), Rous sarcoma virus (RSV), mouse mammary tumor virus (MMTV), human T cell leukemia virus 1 (HTLV-1) and other retroviruses . With few exceptions, LTR-retroelements utilize host tRNAs to prime reverse transcription of their RNA into DNA within viral particles (infectious) or virus-like particles (endogenous) before integrating into the genome. ERVs are endogenous Retroviridae that reside in vertebrates. They are particularly abundant in mammals and are actively mutagenizing mouse genomes .
2. Introduction
Reverse transcription and long terminal repeat (LTR) retroelements are ancient components of eukaryotic genomes [1]. In fact, reverse transcriptase (RT) is one of the most abundant genes in organisms with high copy numbers of retroelements such as mammals [1][2][3]. LTR-retroviruses encode envelope proteins to form virus particles and infect neighboring cells or other organisms, while LTR-retrotransposons that lack functional envelope proteins replicate within viral-like particles (VLPs) to integrate into the same cell. The majority of LTR-retrotransposons in mammals are closely related to known infectious LTR-retroviruses and are therefore called endogenous retroviruses (ERVs). Based on the phylogenetic relationship of their RT genes, mammalian ERVs belong to the Retroviridae genus, while LTR-retrotransposons prevalent in other phyla such as the Gypsy and Copia superfamilies are Metaviridae and Pseudoviridae, respectively [4][5][6]. All three genera include infectious, viral elements with an envelope gene as well as endogenous transposons that proliferate in a strictly intracellular fashion. Endogenous LTR-retrotransposons that lost a functional envelope gene are inherited vertically but may in principle, at low frequency, spread to other organisms by horizontal transfer, a process used by all transposable elements to enter new host species [7][8]. ERVs have become resident aliens in mammalian genomes and many of them were co-opted by their hosts to fulfill essential cellular functions, for example, during placentation and imprinting [9][10][11]. ERV promoter and enhancer activities as well as their protein domains have been useful building blocks during evolution, while their repetitive ends induce recombination and mobility of intact, full-length elements is highly mutagenic [12][13][14][15][16][17]. Hence, their expression needs to be carefully monitored by the cell.
With few exceptions, LTR-retroelements use host tRNA to prime reverse transcription and copy their RNA into DNA for insertion into the genome (Figure 1) [18][19][20][21]. Retroviral proteins bind specific tRNAs with high affinity and recruit them to the virus particle or VLP where their 3′-end initiates reverse transcription at the tRNA primer binding site (PBS). Small RNAs derived from the 3′-end of tRNAs (3′-tRFs) target LTR-retroelements at the PBS and control their mobility and expression [22][23][24]. These highly conserved sequence motifs are a prerequisite for replication and allow host defense mechanisms to identify active LTR-retroelements. ERV sequences make up ~10% of the mouse and human genome. While they are no longer mobile in humans, a number of murine ERVs are highly active and retrotranspose themselves as well as non-autonomous, non-coding family members [and 2 = Lander].
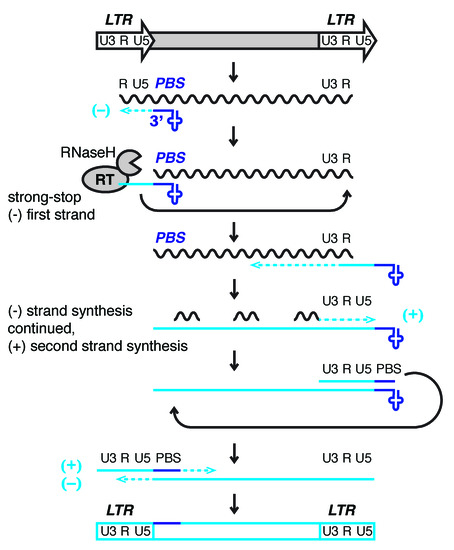
Figure 1. Model of reverse transcription of long terminal repeat (LTR)-retrotransposons and -viruses. LTRs encode promoter elements and termination signals. The RNA transcript contains a region repeated at either end (R), a 5′ unique segment (U5), and a segment only included at the 3′-end of the RNA (U3). The 3′-end of cellular tRNAs (blue cloverleaf) primes reverse transcription by hybridizing to the primer binding site (PBS). While this segment is being copied into first-strand cDNA (light blue line), also called minus (−) strong stop DNA, the RNaseH activity of reverse transcriptase (RT) degrades the template RNA. The elongating cDNA is transferred to the 3′-end of the retrotransposon transcript hybridizing to the R region. The remaining RNA is partially degraded by RNaseH leaving behind primers for second-strand, plus (+) cDNA synthesis. In Retroviridae, the plus strand PBS is a copy of the tRNA primer, while the minus strand is a copy of the original PBS sequence. After another transfer event, first (−) and second (+) strand synthesis are completed to result in a full-length, double-stranded retroviral DNA that will be integrated into the host genome.
3. Epigenetic Reprogramming of ERVs
3.1. De-repression of ERVs during Chromatin Reprogramming
ERVs are usually embedded in repressive heterochromatin, but importantly become active during epigenetic reprogramming in development and disease. Mammals undergo genome-wide epigenetic reprogramming in the embryo right after fertilization and in the germline to obtain totipotency and set aside cells for the next generation [25][26][27]. ERV transcription is repressed by DNA and histone methylation. The histone methyltransferases G9a/GLP, SETDB1, EZH2, histone demethylase KDM1A, as well as the de novo DNA methyltransferases DNMT3a/b, DNMT3L, and DNMT3C establish heterochromatin at different classes of ERVs as discussed in detail elsewhere [28][29]. DNA methylation status can directly correlate with ERV transcription [29][30][31][32][33]. However, absence of DNA methylation does not necessarily lead to ERV expression, as long as histone H3 lysine K9 tri-methylation (H3K9me3) can be maintained [34][35][36][37]. The histone H3K9me3 methyltransferase SETDB1 is acting in complex with KAP1/TRIM28 on fully methylated or fully unmethylated DNA, but not hemi-methylated DNA that is occupied by NP95 [37]. This finding resolves why ERV expression is not always observed in stable methylation knock-outs but is observed in inducible knock-outs that undergo temporary hemi-methylation, and most importantly during epigenetic reprogramming in vivo which includes a hemi-methylated state [32][33][37]. Like any gene, transposon expression depends on multiple layers of repressive and permissive control on the RNA, DNA, and protein level. Removal of silent chromatin marks allows transcription factors (TFs) to bind DNA and promote or inhibit ERV transcription [38][39][40]. LTR sequences, for example, contain species-specific TF binding sites that promote temporary expression of ERVs and neighboring genomic sequences during development [38]. After reprogramming, chromatin patterns at transposable elements need to be re-established through DNA and RNA recognition. KRAB zinc finger proteins (ZFPs) have co-evolved with their transposon targets and guide heterochromatin formation by SETDB1, TRIM28/KAP1 through binding to highly conserved DNA sequence motifs in ERVs [41]. For example, some KRAB-ZFPs bind to the PBS of select ERVs or the polypurine tract that primes second strand reverse transcription during ERV replication [42].
Once transcribed, ERV expression, translation, and reverse transcription must be restricted by the cell, and small RNAs have the ability to recognize and target transposon RNA for silencing. PIWI-interacting RNAs (piRNAs) guide silencing of ERVs in the male germline of mammals [72][73][74]. In the female germline of Muridae, endogenous small interfering RNAs (endo-siRNAs) target transposon mRNA and protect oocytes [26][79][80][81]. Small RNAs derived from the 3′-end of mature tRNAs (3′-tRFs) are expressed in pre-implantation mouse stem cells and potentially protect tissues by targeting the highly conserved tRNA primer binding site (PBS) of ERVs [22]. Small RNA-mediated silencing does not only prevent mutagenic damage from transposition, but importantly regulates repetitive elements that have been co-opted by the host to serve essential functions. For example, silencing of the paternally imprinted Rasgrf1 locus in mouse is mediated by piRNAs that target an ERV sequence [43], and a micro RNA (miRNA) regulates the retrotransposon-like 1 (Rtl1) imprinted gene in mouse placenta [44].
3.2. ERVs as Epigenetic Switches in Development and Disease
The propensity of ERVs to attract diverse silencing machineries that act upon specific transposon families at different stages of development make them ideal epigenetic switches [17]. An estimated 6–30% of transcripts in mouse and human embryonic and somatic tissues are driven by retrotransposon promoters in a highly tissue-specific manner [45]. ERV families define gene-regulatory networks throughout development [12][13]. Transcription of murine MERV-L elements marks the totipotent two-cell stage in early embryos [46]. Human HERV-H expression is indicative of the naive embryonic stem cell state and essential for pluripotency [47][48]. In addition, ERV LTR promoter-enhancer activity drives non-coding, stem-cell specific transcripts that maintain the undifferentiated state and are crucial for cell identity [49][50][51][52]. More than 800 LTRs from the ERV-L and mammalian apparent LTR-retrotransposon (MaLR) families act as alternative promoters and first exons to drive stage-specific gene expression in mammalian oocytes and the developing zygote [25][53]. Taken together, temporary release of transposon silencing during reprogramming affects the transcriptome through (i) expression of potentially mobile, mutagenic, intact transposons, (ii) expression of transposon-derived, long non-coding regulatory RNAs (lncRNAs), and (iii) expression of neighboring genes or lncRNAs driven by promoter-enhancer activities of the LTRs.
The epigenetic state of ERVs and transposable elements in general can not only lead to developmental stage- and cell-type-specific expression but also establish epigenetic alleles or “epialleles” that result in differential expression between isogenic offspring [54][55]. Epialleles can be stable and inherited if they entirely escape reprogramming or “metastable” and lead to stochastic changes of the epigenetic state in the offspring [55]. The most famous example of an ERV-induced metastable epiallele is the differential methylation of an intracisternal A-particle (IAP) insertion upstream the mouse Agouti gene which results in varying fur color and obesity in siblings [30]. In fact, such metastable epialleles of IAP are extremely abundant genome-wide, but few of them affect neighboring gene expression [31]. Select ERVs, particularly a set of IAP elements, are protected from reprogramming in the early embryo and the germline, and therefore inherit their epigenetic state as stable epialleles [56][57]. Human ERV (HERV) methylation varies between individuals that could be metastable epialleles, but it is hard to exclude genetic variation [55][58]. Notably, many imprinted genes are derived from LTR-retrotransposons. Imprinted genes of the sushi-ichi-related retrotransposon homologs (SIRH) are common to placental mammals and derived from Metaviridae gypsy-elements [59]. Lineage-specific Retroviridae ERV insertions mediate imprint establishment at murine loci such as retrotransposon-like 1 (Rtl1), Rasgrf1, Impact, and Slc38a4 [60][44][43]. The murine ERVK family drives non-canonical, histone-dependent imprinting in the extraembryonic lineage [61]. Imprinted loci are established during epigenetic reprogramming of the germline and persist in the early embryo [56][62][63]. In contrast to epialleles, heterochromatin induction at imprinted loci is not stochastic but established at either the paternal or maternal allele, respectively, and is essential for proper development.
Similar to epigenetic reprogramming in development, ERV reactivation has been observed in other tissues with high epigenetic plasticity, particularly in the course of disease [17][64][65]. The role of ERVs in cancer extends beyond their value as diagnostic markers for aberrant reprogramming. They are frequently epigenetically reactivated as cryptic promoters in cancer and drive oncogene expression [49][66][67][68]. Indeed, LTR-retroviruses were originally identified as the causative agents of transmissible tumors in chicken, mice, and humans [69]. Those ‘RNA tumor viruses’ include Rous sarcoma virus (RSV), mouse mammary tumor virus (MMTV), and human T cell leukemia virus 1 (HTLV-1). However, expression of endogenous HERV proteins can also tip the scales and trigger an immune response that drives tumor cells into apoptosis [70].
This entry is adapted from the peer-reviewed paper 10.3390/v12080792
References
- Boeke, J.D. The unusual phylogenetic distribution of retrotransposons: A hypothesis. Genome Res. 2003, 13, 1975–1983, doi:10.1101/gr.1392003.
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921, doi:10.1038/35057062.
- Waterston, R.H.; Lindblad-Toh, K.; Birney, E.; Rogers, J.; Abril, J.F.; Agarwal, P.; Agarwala, R.; Ainscough, R.; Alexandersson, M.; An, P.; et al. Initial sequencing and comparative analysis of the mouse genome. Nature 2002, 420, 520–562, doi:10.1038/nature01262.
- Xiong, Y.; Eickbush, T.H. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 1990, 9, 3353–3362.
- King, A.M.Q.; Lefkowitz, E.J.; Mushegian, A.R.; Adams, M.J.; Dutilh, B.E.; Gorbalenya, A.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; Knowles, N.J., et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2018). Arch. Virol. 2018, 163, 2601–2631, doi:10.1007/s00705-018-3847-1.
- Menendez-Arias, L.; Sebastian-Martin, A.; Alvarez, M. Viral reverse transcriptases. Virus Res. 2017, 234, 153–176, doi:10.1016/j.virusres.2016.12.019.
- Plasterk, R.H.; Izsvak, Z.; Ivics, Z. Resident aliens: The Tc1/mariner superfamily of transposable elements. Trends Genet. 1999, 15, 326–332, doi:10.1016/s0168-9525(99)01777-1.
- Zhang, H.H.; Peccoud, J.; Xu, M.R.; Zhang, X.G.; Gilbert, C. Horizontal transfer and evolution of transposable elements in vertebrates. Nat. Commun. 2020, 11, 1362, doi:10.1038/s41467-020-15149-4.
- Cosby, R.L.; Chang, N.C.; Feschotte, C. Host-transposon interactions: Conflict, cooperation, and cooption. Genes Dev. 2019, 33, 1098–1116, doi:10.1101/gad.327312.119.
- Dupressoir, A.; Vernochet, C.; Bawa, O.; Harper, F.; Pierron, G.; Opolon, P.; Heidmann, T. Syncytin-A knockout mice demonstrate the critical role in placentation of a fusogenic, endogenous retrovirus-derived, envelope gene. Proc. Natl. Acad. Sci. USA 2009, 106, 12127–12132, doi:10.1073/pnas.0902925106.
- Hemberger, M.; Hanna, C.W.; Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 2020, 21, 27–43, doi:10.1038/s41576-019-0169-4.
- Rebollo, R.; Romanish, M.T.; Mager, D.L. Transposable elements: An abundant and natural source of regulatory sequences for host genes. Annu. Rev. Genet. 2012, 46, 21–42, doi:10.1146/annurev-genet-110711-155621.
- Thompson, P.J.; Macfarlan, T.S.; Lorincz, M.C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 2016, 62, 766–776, doi:10.1016/j.molcel.2016.03.029.
- Nellaker, C.; Keane, T.M.; Yalcin, B.; Wong, K.; Agam, A.; Belgard, T.G.; Flint, J.; Adams, D.J.; Frankel, W.N.; Ponting, C.P. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012, 13, R45, doi:10.1186/gb-2012-13-6-r45.
- Thomas, J.; Perron, H.; Feschotte, C. Variation in proviral content among human genomes mediated by LTR recombination. Mob. DNA 2018, 9, 36, doi:10.1186/s13100-018-0142-3.
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296, doi:10.1038/nrg3199.
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86, doi:10.1038/nrg.2016.139.
- Le Grice, S.F. "In the beginning": Initiation of minus strand DNA synthesis in retroviruses and LTR-containing retrotransposons. Biochemistry 2003, 42, 14349–14355, doi:10.1021/bi030201q.
- Levin, H.L. A novel mechanism of self-primed reverse transcription defines a new family of retroelements. Mol. Cell Biol. 1995, 15, 3310–3317.
- Schorn, A.J.; Martienssen, R. Tie-Break: Host and Retrotransposons Play tRNA. Trends Cell Biol. 2018, 28, 793–806, doi:10.1016/j.tcb.2018.05.006.
- Chapman, K.B.; Bystrom, A.S.; Boeke, J.D. Initiator methionine tRNA is essential for Ty1 transposition in yeast. Proc. Natl. Acad. Sci. USA 1992, 89, 3236–3240, doi:10.1073/pnas.89.8.3236.
- Schorn, A.J.; Gutbrod, M.J.; LeBlanc, C.; Martienssen, R. LTR-Retrotransposon Control by tRNA-Derived Small RNAs. Cell 2017, 170, 61–71 e11, doi:10.1016/j.cell.2017.06.013.
- Li, Z.; Ender, C.; Meister, G.; Moore, P.S.; Chang, Y.; John, B. Extensive terminal and asymmetric processing of small RNAs from rRNAs, snoRNAs, snRNAs, and tRNAs. Nucleic Acids Res. 2012, 40, 6787–6799, doi:10.1093/nar/gks307.
- Yeung, M.L.; Bennasser, Y.; Watashi, K.; Le, S.Y.; Houzet, L.; Jeang, K.T. Pyrosequencing of small non-coding RNAs in HIV-1 infected cells: Evidence for the processing of a viral-cellular double-stranded RNA hybrid. Nucleic Acids Res. 2009, 37, 6575–6586, doi:10.1093/nar/gkp707.
- Peaston, A.E.; Evsikov, A.V.; Graber, J.H.; de Vries, W.N.; Holbrook, A.E.; Solter, D.; Knowles, B.B. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev. Cell 2004, 7, 597–606, doi:10.1016/j.devcel.2004.09.004.
- Ohnishi, Y.; Totoki, Y.; Toyoda, A.; Watanabe, T.; Yamamoto, Y.; Tokunaga, K.; Sakaki, Y.; Sasaki, H.; Hohjoh, H. Small RNA class transition from siRNA/piRNA to miRNA during pre-implantation mouse development. Nucleic Acids Res. 2010, 38, 5141–5151, doi:10.1093/nar/gkq229.
- Sasaki, H.; Matsui, Y. Epigenetic events in mammalian germ-cell development: Reprogramming and beyond. Nat. Rev. Genet. 2008, 9, 129–140, doi:10.1038/nrg2295.
- Leung, D.C.; Lorincz, M.C. Silencing of endogenous retroviruses: When and why do histone marks predominate? Trends Biochem. Sci. 2012, 37, 127–133, doi:10.1016/j.tibs.2011.11.006.
- Barau, J.; Teissandier, A.; Zamudio, N.; Roy, S.; Nalesso, V.; Herault, Y.; Guillou, F.; Bourc'his, D. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 2016, 354, 909–912, doi:10.1126/science.aah5143.
- Morgan, H.D.; Sutherland, H.G.; Martin, D.I.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318, doi:10.1038/15490.
- Kazachenka, A.; Bertozzi, T.M.; Sjoberg-Herrera, M.K.; Walker, N.; Gardner, J.; Gunning, R.; Pahita, E.; Adams, S.; Adams, D.; Ferguson-Smith, A.C. Identification, Characterization, and Heritability of Murine Metastable Epialleles: Implications for Non-genetic Inheritance. Cell 2018, 175, 1717, doi:10.1016/j.cell.2018.11.017.
- Walsh, C.P.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117, doi:10.1038/2413.
- Berrens, R.V.; Andrews, S.; Spensberger, D.; Santos, F.; Dean, W.; Gould, P.; Sharif, J.; Olova, N.; Chandra, T.; Koseki, H., et al. An endosiRNA-Based Repression Mechanism Counteracts Transposon Activation during Global DNA Demethylation in Embryonic Stem Cells. Cell Stem Cell 2017, 21, 694–703 e697, doi:10.1016/j.stem.2017.10.004.
- Walter, M.; Teissandier, A.; Perez-Palacios, R.; Bourc'his, D. An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. Elife 2016, 5, doi:10.7554/eLife.11418.
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M., et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687, doi:10.1016/j.stem.2011.04.004.
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931, doi:10.1038/nature08858.
- Sharif, J.; Endo, T.A.; Nakayama, M.; Karimi, M.M.; Shimada, M.; Katsuyama, K.; Goyal, P.; Brind'Amour, J.; Sun, M.A.; Sun, Z., et al. Activation of Endogenous Retroviruses in Dnmt1(-/-) ESCs Involves Disruption of SETDB1-Mediated Repression by NP95 Binding to Hemimethylated DNA. Cell Stem Cell 2016, 19, 81–94, doi:10.1016/j.stem.2016.03.013.
- Robbez-Masson, L.; Rowe, H.M. Retrotransposons shape species-specific embryonic stem cell gene expression. Retrovirology 2015, 12, 45, doi:10.1186/s12977-015-0173-5.
- Friedli, M.; Trono, D. The developmental control of transposable elements and the evolution of higher species. Annu Rev. Cell Dev. Biol. 2015, 31, 429–451, doi:10.1146/annurev-cellbio-100814-125514.
- Dewannieux, M.; Heidmann, T. Endogenous retroviruses: Acquisition, amplification and taming of genome invaders. Curr. Opin. Virol. 2013, 3, 646–656, doi:10.1016/j.coviro.2013.08.005.
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554, doi:10.1038/nature21683.
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623, doi:10.1016/j.devcel.2016.02.024.
- Watanabe, T.; Tomizawa, S.; Mitsuya, K.; Totoki, Y.; Yamamoto, Y.; Kuramochi-Miyagawa, S.; Iida, N.; Hoki, Y.; Murphy, P.J.; Toyoda, A., et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 2011, 332, 848–852, doi:10.1126/science.1203919.
- Ito, M.; Sferruzzi-Perri, A.N.; Edwards, C.A.; Adalsteinsson, B.T.; Allen, S.E.; Loo, T.H.; Kitazawa, M.; Kaneko-Ishino, T.; Ishino, F.; Stewart, C.L., et al. A trans-homologue interaction between reciprocally imprinted miR-127 and Rtl1 regulates placenta development. Development 2015, 142, 2425–2430, doi:10.1242/dev.121996.
- Faulkner, G.J.; Kimura, Y.; Daub, C.O.; Wani, S.; Plessy, C.; Irvine, K.M.; Schroder, K.; Cloonan, N.; Steptoe, A.L.; Lassmann, T., et al. The regulated retrotransposon transcriptome of mammalian cells. Nat. Genet. 2009, 41, 563–571, doi:10.1038/ng.368.
- Macfarlan, T.S.; Gifford, W.D.; Driscoll, S.; Lettieri, K.; Rowe, H.M.; Bonanomi, D.; Firth, A.; Singer, O.; Trono, D.; Pfaff, S.L. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 2012, 487, 57–63, doi:10.1038/nature11244.
- Lu, X.; Sachs, F.; Ramsay, L.; Jacques, P.E.; Goke, J.; Bourque, G.; Ng, H.H. The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat. Struct. Mol. Biol. 2014, 21, 423–425, doi:10.1038/nsmb.2799.
- Wang, J.; Xie, G.; Singh, M.; Ghanbarian, A.T.; Rasko, T.; Szvetnik, A.; Cai, H.; Besser, D.; Prigione, A.; Fuchs, N.V., et al. Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 2014, 516, 405–409, doi:10.1038/nature13804.
- Herquel, B.; Ouararhni, K.; Martianov, I.; Le Gras, S.; Ye, T.; Keime, C.; Lerouge, T.; Jost, B.; Cammas, F.; Losson, R., et al. Trim24-repressed VL30 retrotransposons regulate gene expression by producing noncoding RNA. Nat. Struct. Mol. Biol. 2013, 20, 339–346, doi:10.1038/nsmb.2496.
- Fort, A.; Hashimoto, K.; Yamada, D.; Salimullah, M.; Keya, C.A.; Saxena, A.; Bonetti, A.; Voineagu, I.; Bertin, N.; Kratz, A., et al. Deep transcriptome profiling of mammalian stem cells supports a regulatory role for retrotransposons in pluripotency maintenance. Nat. Genet. 2014, 46, 558–566, doi:10.1038/ng.2965.
- Pontis, J.; Planet, E.; Offner, S.; Turelli, P.; Duc, J.; Coudray, A.; Theunissen, T.W.; Jaenisch, R.; Trono, D. Hominoid-Specific Transposable Elements and KZFPs Facilitate Human Embryonic Genome Activation and Control Transcription in Naive Human ESCs. Cell Stem Cell 2019, 24, 724–735 e725, doi:10.1016/j.stem.2019.03.012.
- Kunarso, G.; Chia, N.Y.; Jeyakani, J.; Hwang, C.; Lu, X.; Chan, Y.S.; Ng, H.H.; Bourque, G. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat. Genet. 2010, 42, 631–634, doi:10.1038/ng.600.
- Franke, V.; Ganesh, S.; Karlic, R.; Malik, R.; Pasulka, J.; Horvat, F.; Kuzman, M.; Fulka, H.; Cernohorska, M.; Urbanova, J., et al. Long terminal repeats power evolution of genes and gene expression programs in mammalian oocytes and zygotes. Genome Res. 2017, 27, 1384–1394, doi:10.1101/gr.216150.116.
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109, doi:10.1016/j.cell.2014.02.045.
- Bertozzi, T.M.; Ferguson-Smith, A.C. Metastable epialleles and their contribution to epigenetic inheritance in mammals. Semin Cell Dev. Biol. 2020, 97, 93–105, doi:10.1016/j.semcdb.2019.08.002.
- Seisenberger, S.; Andrews, S.; Krueger, F.; Arand, J.; Walter, J.; Santos, F.; Popp, C.; Thienpont, B.; Dean, W.; Reik, W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell 2012, 48, 849–862, doi:10.1016/j.molcel.2012.11.001.
- Nakamura, T.; Arai, Y.; Umehara, H.; Masuhara, M.; Kimura, T.; Taniguchi, H.; Sekimoto, T.; Ikawa, M.; Yoneda, Y.; Okabe, M., et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat. Cell Biol. 2007, 9, 64–71, doi:10.1038/ncb1519.
- Reiss, D.; Mager, D.L. Stochastic epigenetic silencing of retrotransposons: Does stability come with age? Gene 2007, 390, 130–135, doi:10.1016/j.gene.2006.07.032.
- Kaneko-Ishino, T.; Ishino, F. The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front. Microbiol. 2012, 3, 262, doi:10.3389/fmicb.2012.00262.
- Bogutz, A.B.; Brind'Amour, J.; Kobayashi, H.; Jensen, K.N.; Nakabayashi, K.; Imai, H.; Lorincz, M.C.; Lefebvre, L. Evolution of imprinting via lineage-specific insertion of retroviral promoters. Nat. Commun. 2019, 10, 5674, doi:10.1038/s41467-019-13662-9.
- Hanna, C.W.; Perez-Palacios, R.; Gahurova, L.; Schubert, M.; Krueger, F.; Biggins, L.; Andrews, S.; Colome-Tatche, M.; Bourc'his, D.; Dean, W., et al. Endogenous retroviral insertions drive non-canonical imprinting in extra-embryonic tissues. Genome Biol. 2019, 20, 225, doi:10.1186/s13059-019-1833-x.
- Mackin, S.J.; Thakur, A.; Walsh, C.P. Imprint stability and plasticity during development. Reproduction 2018, 156, R43-R55, doi:10.1530/REP-18-0051.
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452, doi:10.1126/science.1229277.
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Consortium, N.A.; Fagegaltier, D.; Harris, B.T., et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177 e1165, doi:10.1016/j.celrep.2019.09.066.
- Krug, L.; Chatterjee, N.; Borges-Monroy, R.; Hearn, S.; Liao, W.W.; Morrill, K.; Prazak, L.; Rozhkov, N.; Theodorou, D.; Hammell, M., et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017, 13, e1006635, doi:10.1371/journal.pgen.1006635.
- Jang, H.S.; Shah, N.M.; Du, A.Y.; Dailey, Z.Z.; Pehrsson, E.C.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S., et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 2019, 51, 611–617, doi:10.1038/s41588-019-0373-3.
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424, doi:10.1038/nrc.2017.35.
- Babaian, A.; Mager, D.L. Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 2016, 7, 24, doi:10.1186/s13100-016-0080-x.
- Weiss, R.T., N.; Varmus, H.; Coffin, J. RNA Tumor Viruses; Cold Spring Harbor Laboratory Press: Plainview, NY, USA, 1982.
- Bannert, N.; Hofmann, H.; Block, A.; Hohn, O. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front. Microbiol. 2018, 9, 178, doi:10.3389/fmicb.2018.00178.
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285, doi:10.1038/nrg2072.
- Siomi, M.C.; Sato, K.; Pezic, D.; Aravin, A.A. PIWI-interacting small RNAs: The vanguard of genome defence. Nat. Rev. Mol. Cell Biol. 2011, 12, 246–258, doi:10.1038/nrm3089.
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O'Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108, doi:10.1038/s41576-018-0073-3.
- Ernst, C.; Odom, D.T.; Kutter, C. The emergence of piRNAs against transposon invasion to preserve mammalian genome integrity. Nat. Commun. 2017, 8, 1411, doi:10.1038/s41467-017-01049-7.
- Aravin, A.A.; Sachidanandam, R.; Bourc'his, D.; Schaefer, C.; Pezic, D.; Toth, K.F.; Bestor, T.; Hannon, G.J. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 2008, 31, 785–799, doi:10.1016/j.molcel.2008.09.003.
- Kuramochi-Miyagawa, S.; Watanabe, T.; Gotoh, K.; Totoki, Y.; Toyoda, A.; Ikawa, M.; Asada, N.; Kojima, K.; Yamaguchi, Y.; Ijiri, T.W., et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 2008, 22, 908–917, doi:10.1101/gad.1640708.
- Carmell, M.A.; Girard, A.; van de Kant, H.J.; Bourc'his, D.; Bestor, T.H.; de Rooij, D.G.; Hannon, G.J. MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev. Cell 2007, 12, 503–514, doi:10.1016/j.devcel.2007.03.001.
- Castel, S.E.; Martienssen, R.A. RNA interference in the nucleus: Roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 2013, 14, 100–112, doi:10.1038/nrg3355.
- Juliano, C.; Wang, J.; Lin, H. Uniting germline and stem cells: The function of Piwi proteins and the piRNA pathway in diverse organisms. Annu. Rev. Genet. 2011, 45, 447–469, doi:10.1146/annurev-genet-110410-132541.
- Tam, O.H.; Aravin, A.A.; Stein, P.; Girard, A.; Murchison, E.P.; Cheloufi, S.; Hodges, E.; Anger, M.; Sachidanandam, R.; Schultz, R.M., et al. Pseudogene-derived small interfering RNAs regulate gene expression in mouse oocytes. Nature 2008, 453, 534–538, doi:10.1038/nature06904.
- Watanabe, T.; Totoki, Y.; Toyoda, A.; Kaneda, M.; Kuramochi-Miyagawa, S.; Obata, Y.; Chiba, H.; Kohara, Y.; Kono, T.; Nakano, T., et al. Endogenous siRNAs from naturally formed dsRNAs regulate transcripts in mouse oocytes. Nature 2008, 453, 539–543, doi:10.1038/nature06908.