Fabry disease (FD) is a lysosomal storage disorder, depending on defects in alpha-galactosidase A (GAL) activity. At the clinical level, FD shows a high phenotype variability. Cardiovascular dysfunction is often recurrent or, in some cases, is the sole symptom (cardiac variant) representing the leading cause of death in Fabry patients. Cardiac dysfunction is dependent on globotriaosylceramide (Gb3) accumulation in the heart but several other mechanisms are involved, such as inflammation and mitochondrial dysfunction, that could become useful targets for therapeutics.
- fabry
- lysosomal disorder
- cardiovascular disease
1. Introduction
Lysosomal storage disorders comprise a group of rare inherited metabolic diseases characterized by abnormal deposition of intracellular wasting materials due to enzyme deficiencies. Among them, Fabry disease (FD) is an X-linked inherited disorder of glycosphingolipid metabolism due to deficient or absent lysosomal alpha-galactosidase A (GAL) activity, which results in a progressive accumulation of globotriaosylceramide (Gb3) and its metabolites [1][2]. Such a mechanism is considered responsible for the damage of several organs, including the kidney, heart, lung, small intestine, brain, and liver, thus allowing FD classification as a “multiorgan” disorder. The diagnostic signature is the detection of Gb3 in urine and plasma, which is confirmed by genetic analysis. In some FD patients, especially females, urine Gb3 are not altered, limiting its use as a diagnostic tool [3][4]. Recently, lyso-Gb3, a deacylated analogue of Gb3, emerged as a novel indicator of FD, showing a greater sensitivity than Gb3 in FD females [4]. Although Fabry patients share the same pathogenetic mechanism (defect in GAL activity and accumulation of Gb3 in tissues), FD appears a “multifaced” disorder, and patients show significant variability in clinical signs. According to the canonic classification, Fabry phenotypes categorize into two specific groups classic and non-classic. The classic phenotype includes the manifestation of multiple symptoms during childhood or adolescence with males affected earlier than females. The main symptoms comprise neurological pain, acroparesthesia, and episodic “Fabry crises” of acute pain as well as significant renal, cardiac, and cerebrovascular complications that could manifest in later stages. In the non-classic phenotype, the clinical signs have mainly a later onset (fourth to the sixth decade of life), but they could manifest in childhood with very different symptoms from those of the classic phenotype. Adult-onset cardiac (cardiomegaly, left ventricular hypertrophy, cardiomyopathy, hypertrophic cardiomyopathy, and myocardial infarction) and renal (end-stage renal disease) variants are more prevalent. Although FD is an X-linked disorder, heterozygous females have symptoms ranging from very mild to severe due to random X-chromosome inactivation [5]. Thus, FD is a very complex condition due to the significant variability of clinical phenotypes.
2. The Cardiac Phenotype
Cardiovascular diseases are the leading cause of death in Fabry patients; left ventricular hypertrophy (LVH) and myocardial fibrosis are the main risk factors for death [6]. LVH is the most common clinical sign shown by echocardiography in several Fabry patients [7]. Hypertrophy starts with concentric remodelling, which progressively evolves to overt hypertrophy with cardiac fibrosis and reduced contractile performance [8]. However, hypertrophy could be not revealed in the early stages of the disease. At the same time, echocardiography could detect diastolic dysfunction [9], suggesting that alterations of diastolic functions could precede cardiac hypertrophy. Diastolic dysfunction is a common feature of Fabry disease while left ventricular systolic dysfunction reduces ejection fraction or fractional shortening rarely occurs [8]. A high frequency of ischemic events and myocardial infarctions is also detected in Fabry patients. A reduced myocardial perfusion reserve characterizes Fabry disease while the peripheral artery endothelial function is preserved [10]. Electrophysiological abnormalities and arrhythmias could also occur [11][12][13]. The most frequent rhythm abnormalities include supraventricular tachycardias and atrial fibrillation and flutter. Valvular structural abnormalities are frequent, especially in the left heart valves, due to valvular infiltration [14]. Advances in the knowledge of FD’s cardiac phenotype are due to cardiac magnetic resonance (CMR). CMR allows a non-invasive tissue characterization, including the assessment of myocardial fibrosis, through late gadolinium enhancement, and sphingolipid storage, by T1 mapping. A study from Nordin and colleagues clarified the developmental stages of cardiac phenotype in FD patients based on these parameters [15]. This phenotype’s evolution starts with an accumulation phase that occurs in childhood and is characterized by ECG changes and low T1. Myocyte hypertrophy and inflammation phase follows with chronic troponin increase, low T1, and left ventricle posterior wall thinning, especially in women. The latest step is characterized by persistent LVH and troponin increase, fibrosis, NT-proBNP elevation, and clinical heart failure [15].
The cardiovascular phenotype in FD is not shown in all patients and, when it occurs, appears with variable severity. A possible explanation could be different types of mutations since some seem to be more strictly associated with cardiac disease. Ile239Met mutation in the GLA gene occurs in a family with a predominant cardiac phenotype of Fabry disease [16]. Individuals from this family carrying Ile239Met mutation display LVH and were mainly females [16]. Furthermore, the p.F113L mutation of the GLA gene seems to cause the late-onset phenotype of FD with predominant cardiac manifestations [17]. However, multiple mutations could occur in the same individual, which allow the development of LVH. Thus, the involvement of other pathogenetic mechanisms in cardiac dysfunction development, as described below, should also be considered.
2.1 The Cardiac Variant
There are some rare cases, especially in male hemizygotes, in which cardiac hypertrophy is the only overt clinical sign of FD [57]. These patients do not show the typical symptoms of FD but develop heart failure. These cases are defined as “cardiac variants” and are characterized by a Gb3 accumulation exclusively in the heart, causing cardiac damage without affecting the other organs. Several findings suggest that this phenomenon is probably due to residual alpha-galactosidase A activity, which delays the disease’s progression, thus preventing the early manifestation of clinical signs. [58]. Since this phenomenon depends on alpha-galactosidase A activity, it could be associated with specific GLA gene mutations.
Even if the cardiac variant occurs mostly in male hemizygotes, a rare case of a woman with FD’s cardiac variant has also been described. The genetic test revealed a missense heterozygous mutation in the GLA gene, c. 395 G/G>G/A, p. G132E, and the urinary sediment showed Mulberry cells’ presence [59]. These data suggest screening all patients with heart failure without typical FD symptoms for urinary Mulberry cells to allow early diagnosis and prompt therapy [59].
2.2. Molecular Mechanisms of Cardiac Dysfunction
How does Gb3 accumulation in the heart alter cardiac function? Several reports propose the involvement of different molecular mechanisms. CM differentiated from a patient-derived iPSC show accumulation of Gb3, GAL activity deficiency, and ANP expression increase [18]. These features are associated with increased excitability, impaired electrophysiology, and altered calcium handling [19] due to increased expression of the membrane Ca2 + sensor L-type calcium channels, hyperphosphorylation ryanodine receptor and decreased expression of phospholamban [19][20]. Such findings suggest that defects in calcium handling could contribute to developing the cardiac phenotype in FD. Cardiac energy metabolism was also shown to be altered in FD [21]. In GLA-null cardiomyocytes, Gb3 accumulation associates with impaired autophagic flux, alteration of mitochondrial membrane potential, and increased ROS production [22][23][24]. This evidence suggests the crosstalk between loss of GAL activity and mitochondria dysfunction in cardiac cells. Given these findings and the known role of mitochondria in the development of heart diseases [25][26][27][28], it is likely to suppose that mitochondrial alterations could play a primary role in the development and progression of the cardiac phenotype in Fabry patients.
Inflammation is also associated with cardiac dysfunction in FD. Indeed, a multicenter cohort study in Fabry and control patients revealed that inflammatory and cardiac remodelling biomarkers are elevated in Fabry patients and correlate with a faster disease progression in patients with more severe clinical features [29]. Furthermore, a clinical study shows that myocarditis is detectable in half of Fabry patients with cardiomyopathy and correlates with disease severity [30]. Giving the known role of inflammation in the development of cardiovascular diseases [31][32], it is believable that the chronic inflammatory state could trigger or worsen cardiac damage in Fabry patients. In this context, anti-inflammatory therapy could become adjuvant to prevent irreversible cardiac damage in FD.
In multiple degenerative and acute diseases, mitochondrial dysfunction and inflammation are strictly linked [33]. Indeed, mitochondrial dysfunction increases ROS production, causing oxidative damage in both the heart and the vasculature, and therefore, inducing vascular inflammation. ROS triggers the NFkappaB pathway leading to activation of NLRP3 inflammasome. The generation of inflammatory cytokines, in turn, causes endothelial and mitochondrial dysfunction by further activating NFkappaB [33]. Furthermore, the activation of NFkappaB is also involved in alterations of endothelial function and the expression of pro-hypertrophic genes [34][35]. Thus, the hypothesis raises that a vicious circle is established in response to Gb3 accumulation, mainly based on NFkappaB activation as an amplifier of processes disruption and cardiac cell damage in FD (Figure 1). If such a hypothesis is confirmed, NFkappaB could be a novel target in FD.
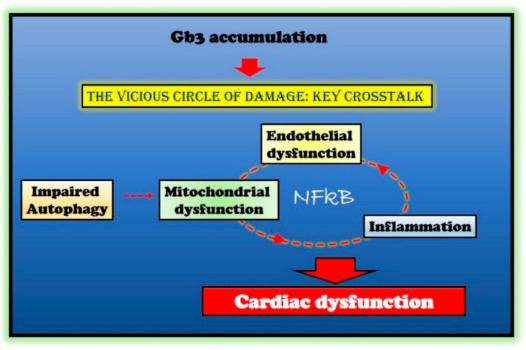
Figure 1. Molecular mechanisms in Fabry disease (FD) cardiac damage. Globotriaosylceramide (Gb3) accumulation in Fabry disease impairs several critical processes within the cell (autophagy and endothelial and mitochondrial function) and induces an inflammatory state. The crosstalk between these impaired conditions is responsible for cardiac damage in Fabry patients. The nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) represents the critical node in this vicious circle of damage.
3. Conclusions
FD is a multifaceted condition with variable clinic phenotypes among patients. The cardiovascular one is the most recurrent and is also the primary cause of death. The main trigger of the dangerous machine is the lack of GAL activity and the accumulation of Gb3 in lysosomes, which are, in fact, the main target of available therapies. However, in the last decade, other important cardiovascular mechanisms have been revealed, such as genetic mutations, endothelial dysfunction, alterations of energetic metabolism, and activation of inflammatory molecules. Among them, inflammation and mitochondrial dysfunction seem to be the most promising possible target, and NFkappaB is a potential common mechanism. Further studies are needed that will allow developing more effective drugs to save and facilitate all patients affected by FD.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22031331
References
- Germain, D. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30.
- Boutin, M.; Menkovic, I.; Martineau, T.; Vaillancourt-Lavigueur, V.; Toupin, A.; Auray-Blais, C. Separation and Analysis of Lactosylceramide, Galabiosylceramide, and Globotriaosylceramide by LC-MS/MS in Urine of Fabry Disease Patients. Anal. Chem. 2017, 89, 13382–13390.
- Young, E.; Mills, K.; Morris, P.; Vellodi, A.; Lee, P.; Waldek, S.; Winchester, B. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatr. Suppl. 2005, 94, 51–54.
- Young-Gqamana, B.; Brignol, N.; Chang, H.-H.; Khanna, R.; Soska, R.; Fuller, M.; Sitaraman, S.A.; Germain, D.P.; Giugliani, R.; Hughes, D.A.; et al. Migalastat HCl Reduces Globotriaosylsphingosine (Lyso-Gb3) in Fabry Transgenic Mice and in the Plasma of Fabry Patients. PLoS ONE 2013, 8, e57631.
- Deegan, P.B.; Baehner, A.F.; Romero, M.-Á.B.; Hughes, D.A.; Kampmann, C.; Beck, M. Natural history of Fabry disease in females in the Fabry Outcome Survey. J. Med. Genet. 2006, 43, 347–352.
- Mehta, A.; Clarke, J.T.; Giugliani, R.; Elliott, P.; Linhart, A.; Beck, M.; Sunder-Plassmann, G.; Investigators, F.O.S. Natural course of Fabry disease: Changing pattern of causes of death in FOS—Fabry Outcome Survey. J. Med. Genet. 2009, 46, 548–552.
- Linhart, A.; Paleček, T.; Bultas, J.; Ferguson, J.J.; Hrudová, J.; Karetová, D.; Zeman, J.; Ledvinová, J.; Poupětová, H.; Elleder, M.; et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am. Heart J. 2000, 139, 1101–1108.
- Linhart, A. The heart in Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; NCBI: Bethesda, MD, USA, 2006.
- Yenercag, M.; Arslan, U. Tp-e interval and Tp-e/QT ratio and their association with left ventricular diastolic dysfunction in Fabry disease without left ventricular hypertrophy. J. Electrocardiol. 2020, 59, 20–24.
- Kalliokoski, R.J.; Kalliokoski, K.; Sundell, J.; Engblom, E.; Penttinen, M.; Kantola, I.; Raitakari, O.; Knuuti, J.; Nuutila, P. Impaired myocardial perfusion reserve but preserved peripheral endothelial function in patients with Fabry disease. J. Inherit. Metab. Dis. 2005, 28, 563–573.
- Pochis, W.T.; Litzow, J.T.; King, B.G.; Kenny, D. Electrophysiologic findings in Fabry’s disease with a short PR interval. Am. J. Cardiol. 1994, 74, 203–204.
- Shah, J.S.; Hughes, D.A.; Sachdev, B.; Tome, M.; Ward, D.; Lee, P.; Mehta, A.B.; Elliott, P.M. Prevalence and Clinical Significance of Cardiac Arrhythmia in Anderson-Fabry Disease. Am. J. Cardiol. 2005, 96, 842–846.
- Shanks, M.; Thompson, R.B.; Paterson, I.D.; Putko, B.; Khan, A.; Chan, A.; Becher, H.; Oudit, G.Y. Systolic and Diastolic Function Assessment in Fabry Disease Patients Using Speckle-Tracking Imaging and Comparison with Conventional Echocardiographic Measurements. J. Am. Soc. Echocardiogr. 2013, 26, 1407–1414.
- Linhart, A.; Lubanda, J.-C.; Palecek, T.; Bultas, J.; Karetová, D.; Ledvinová, J.; Elleder, M.; Aschermann, M. Cardiac manifestations in Fabry disease. J. Inherit. Metab. Dis. 2001, 24, 75–83.
- Nordin, S.; Kozor, R.; Medina-Menacho, K.; Abdel-Gadir, A.; Baig, S.; Sado, D.M.; Lobascio, I.; Murphy, E.; Lachmann, R.H.; Mehta, A.; et al. Proposed Stages of Myocardial Phenotype Development in Fabry Disease. JACC Cardiovasc. Imaging 2019, 12, 1673–1683.
- Csanyi, B.; Hategan, L.; Nagy, V.; Obal, I.; Varga, E.T.; Borbas, J.; Tringer, A.; Eichler, S.; Forster, T.; Rolfs, A.; et al. Identification of a Novel GLA Gene Mutation, p.Ile239Met, in Fabry Disease With a Predominant Cardiac Phenotype. Int. Heart J. 2017, 58, 454–458.
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Robles, A.R.; Costa, M.A.; Pereira, O.; Vide, A.T.; Branco, G.C.; Simões, S.; Guimarães, M.J.; et al. Natural history of the late-onset phenotype of Fabry disease due to the p.F113L mutation. Mol. Genet. Metab. Rep. 2020, 22, 100565.
- Kuramoto, Y.; Naito, A.T.; Tojo, H.; Sakai, T.; Ito, M.; Shibamoto, M.; Nakagawa, A.; Higo, T.; Okada, K.; Yamaguchi, T.; et al. Generation of Fabry cardiomyopathy model for drug screening using induced pluripotent stem cell-derived cardiomyocytes from a female Fabry patient. J. Mol. Cell. Cardiol. 2018, 121, 256–265.
- Birket, M.J.; Raibaud, S.; Lettieri, M.; Adamson, A.D.; Letang, V.; Cervello, P.; Redon, N.; Ret, G.; Viale, S.; Wang, B.; et al. A Human Stem Cell Model of Fabry Disease Implicates LIMP-2 Accumulation in Cardiomyocyte Pathology. Stem Cell Rep. 2019, 13, 380–393.
- Meng, X.; Shen, J.; Kong, M.; Brady, R.; Li, R.A.; Schiffmann, R. Abnormal intracellular calcium handling: A key pathogenic and therapeutic target of the cardiac manifestations in Fabry disease. Mol. Genet. Metab. 2014, 111, S77.
- Machann, W.; Breunig, F.; Weidemann, F.; Sandstede, J.; Hahn, D.; Köstler, H.; Neubauer, S.; Wanner, C.; Beer, M. Cardiac energy metabolism is disturbed in Fabry disease and improves with enzyme replacement therapy using recombinant human galactosidase A. Eur. J. Heart Fail. 2011, 13, 278–283.
- Song, H.-Y.; Chien, C.-S.; Yarmishyn, A.A.; Chou, S.-J.; Yang, Y.-P.; Wang, M.-L.; Leu, H.-B.; Yu, W.-C.; Chang, Y.-L.; Chiou, S.-H. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327.
- Ravarotto, V.; Simioni, F.; Carraro, G.; Bertoldi, G.; Pagnin, E.; Calò, L.A. Oxidative Stress and Cardiovascular-Renal Damage in Fabry Disease: Is There Room for a Pathophysiological Involvement? J. Clin. Med. 2018, 7, 409.
- Ravarotto, V.; Carraro, G.; Pagnin, E.; Bertoldi, G.; Simioni, F.; Maiolino, G.; Martinato, M.; Landini, L.; Davis, P.A.; Calò, L.A. Oxidative stress and the altered reaction to it in Fabry disease: A possible target for cardiovascular-renal remodeling? PLoS ONE 2018, 13, e0204618.
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250.
- Ciccarelli, M.; Sorriento, D.; Fiordelisi, A.; Gambardella, J.; Franco, A.; Del Giudice, C.; Sala, M.; Monti, M.G.; Bertamino, A.; Campiglia, P.; et al. Pharmacological inhibition of GRK2 improves cardiac metabolism and function in experimental heart failure. ESC Heart Fail. 2020, 7, 1571–1584.
- Sorriento, D.; Ciccarelli, M.; Cipolletta, E.; Trimarco, B.; Iaccarino, G. “Freeze, Don’t Move”: How to Arrest a Suspect in Heart Failure—A Review on Available GRK2 Inhibitors. Front. Cardiovasc. Med. 2016, 3, 48.
- Sorriento, D.; Franco, A.; Rusciano, M.R.; Maione, A.S.; Soprano, M.; Illario, M.; Iaccarino, G.; Ciccarelli, M. Good at Heart: Preserving Cardiac Metabolism during aging. Curr. Diabetes Rev. 2015, 12, 90–99.
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients With Fabry Disease: A Critical Link to Heart Failure With Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009098.
- Frustaci, A.; Verardo, R.; Grande, C.; Galea, N.; Piselli, P.; Carbone, I.; Alfarano, M.; Russo, M.A.; Chimenti, C. Immune-Mediated Myocarditis in Fabry Disease Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, e009052.
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a Key Player in the Crosstalk between Inflammation and Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 1599.
- Sorriento, D.; Iaccarino, G. Inflammation and Cardiovascular Diseases: The Most Recent Findings. Int. J. Mol. Sci. 2019, 20, 3879.
- Lopez-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-Garcia, C.; Valcarcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118.
- Sorriento, D.; Ciccarelli, M.; Santulli, G.; Campanile, A.; Altobelli, G.G.; Cimini, V.; Galasso, G.; Astone, D.; Piscione, F.; Pastore, L.; et al. The G-protein-coupled receptor kinase 5 inhibits NFkappaB transcriptional activity by inducing nuclear accumulation of IkappaB alpha. Proc. Natl. Acad. Sci. USA 2008, 105, 17818–17823.
- Sorriento, D.; Santulli, G.; Fusco, A.; Anastasio, A.; Trimarco, B.; Iaccarino, G. Intracardiac injection of AdGRK5-NT reduces left ventricular hypertrophy by inhibiting NF-kappaB-dependent hypertrophic gene expression. Hypertension 2010, 56, 696–704.