Age-related macular degeneration (AMD) is a common irreversible sight-threatening disease characterized by progressive degeneration of the central retina, preferentially involving the retinal photoreceptors, the retinal pigment epithelium (RPE), the Bruch’s membrane (BM), or the choroidal microcirculation in the macular region.
- age-related macular degeneration
- pathogenesis
1. Introduction
Age-related macular degeneration (AMD) is a common irreversible sight-threatening disease characterized by progressive degeneration of the central retina, preferentially involving the retinal photoreceptors, the retinal pigment epithelium (RPE), the Bruch’s membrane (BM), or the choroidal microcirculation in the macular region [1]. In 2020, AMD accounted for 5.4% (1.8 million) of blindness among the global 33.6 million blind adults over the age of 50 and is the fourth leading cause of blindness worldwide [2]. Moreover, the prevalence of AMD is predicted to rise continuously and rapidly based on the increasing average life expectancy [3][4]. It has been estimated that in 2040, 288 million people will be affected by AMD worldwide [5]. Depending on the degree of disease severity, patients perceive different decreases in their quality of life [6]. For example, patients with mild AMD perceive a 17% decrement in their quality of life, while people with severe AMD report on a 63% reduction in the quality of life [6]. In addition to the deleterious effects on patients’ quality of life, treatment of AMD causes high economic costs [6]. The annual loss of gross domestic product due to AMD was $2.6 billion in Canada in 2005, while the annual loss was approximately $4.6 billion in the United States in 2016 [1][6].
The first reports of the pathogenetic process underlying AMD were described by Donders et al. in 1855 and by Nagel et al. in 1868 [7][8]. By 1965, the terminology of AMD was becoming more and more accepted [9]. AMD is classified into three clinical stages: early, intermediate, and advanced AMD [10]. The presence of drusen (>63 and ≤125 μm in diameter) is the earliest clinical feature of early AMD, which impairs the patients’ ability of dark adaptation during the transition from high to low illumination environments [10]. Most central visual loss occurs in the intermediate and advanced stages of AMD. Advanced AMD includes two categories: geographic atrophy (GA) and neovascular AMD [11]. GA is characterized by slowly progressive deterioration of the RPE, photoreceptor layer, and choroidal capillaries in the macula, leading to progressive vision loss over several years [12]. Neovascular AMD, also known as exudative AMD, is characterized by the invasion of new immature choroidal vessels breaking through the BM into the retina, causing exudates, hemorrhages and detachment of the RPE or retina. This disease form causes more rapid progressive loss of vision than GA [12][13].
Many risk factors are related to AMD and interact with each other in its pathogenesis, making AMD a complex multifactorial disease [14]. The multifactorial etiology includes demographic factors (age, gender, and ethnicity), epidemiological risk factors (body-mass index, smoking, diet, and gene polymorphisms, e.g., mutations in the complement cascade), and environmental risk factors (exposure to sunlight and to chemical substances) [15][16][17][18]. Notably, oxidative stress and choroidal vascular dysfunction have been suggested to be the most important trigger factors of AMD pathogenesis [19][20][21]. Oxidative stress plays an important role in aging diseases including AMD, especially owing to the much higher oxygen consumption by the retina than by any other tissue [22]. In addition, the proliferative abnormal choroidal vasculature grows into the subretinal space in the exudative form of AMD, eventually causing detachment of the RPE and consecutively vision loss [23]. It is more and more recognized that genetic risk factors have a critical relevance to the oxidative stress response and choroidal vascular dysfunction in AMD [24][25]. However, still little is known regarding the interplay between these two pathophysiological factors and their link to genetic factors in AMD pathogenesis. As yet, up to 90% of AMD patients worldwide are still untreatable [13]. This situation makes it an urgent priority to better understand the pathophysiology of AMD and to design targeted therapies for this disease.
2. The Macula
The macula is an oval-shaped spot localized in the center of the retina, which is responsible for clear and fine detail vision [26]. The macula has a diameter of about 5 mm and can be subdivided into six areas: the umbo, foveola, foveal avascular zone, fovea, parafovea, and perifovea areas [26]. The fovea is located at the center of the macula and contains the largest concentration of cones in the retina, enabling high-resolution vision [26]. The central region of the macula, which is 250 to 600 μm in diameter and termed the foveal avascular zone, lacks retinal blood vessels and is supplied by the choroidal circulation [27].
The fovea is composed of few layers from anterior to posterior: an extremely thin inner plexiform layer, the outer nuclear layer, the cones, and the RPE layer [28]. The foveola lies in the center of the fovea and contains only cone photoreceptors and unique Müller cells with optical fiber characteristics [29]. Moreover, peripheral areas of the macula and the rest of the retina contain both rod and cone cells [30]. The RPE is attached to the cone photoreceptors and carries out many functions including phagocytosis of the photoreceptors’ outer segment membrane, maintenance of the physiological functions of the choriocapillaris, conversion and storage of retinoid, absorption of scattered light, and transport of ions and fluid [31]. RPE cells are taller in the fovea than in non-foveal areas [32]. The BM is attached to the basal surface of the RPE, an elastic semi-permeable barrier for major metabolic transport and exchange [33]. Adjacent to BM, the choriocapillaris is composed of fenestrated capillaries in the innermost layer of the choroid that provides blood supply to the RPE and the macula (Figure 1) [34]. A better understanding of macular anatomy can significantly improve our understanding regarding the role of risk factors in the pathogenesis of AMD.
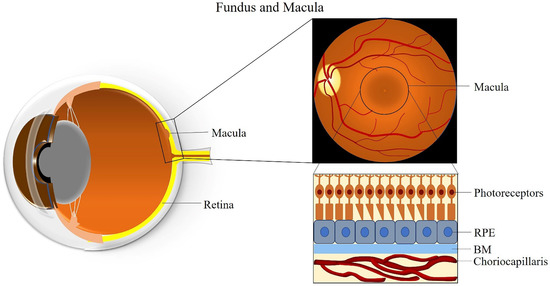
3. Clinical Classification of AMD
AMD is a degenerative disease of the retina, which leads to changes in photoreceptors, RPE, BM, and/or choriocapillaris, eventually resulting in central visual impairment [35][36]. The pathology of AMD is characterized by macular drusen, RPE atrophy, choroidal neovascularization, neurosensory retina detachments, and disciform scars or lesions [37]. According to the clinical manifestation, several classification scales of AMD have been developed. For example, The Age-Related Eye Disease Study (AREDS) research group divided patients into four AMD categories, depending on the size and extent of drusen, presence of GA, and neovascular changes [38]. Later, the AREDS research group developed a nine-step fundus photographic severity scale for AMD, combining the six-step drusen area and five-step pigmentary abnormality area scales for tracking the progression of AMD and providing baseline risk categories [37]. However, the AREDS nine-step severity scale is overly complicated and not useful for clinical work [39]. Consequently, the AREDS research group proposed a five-step simplified clinical scale for AMD, which is clinically more relevant [40].
However, the precise definition for clinical classification of AMD is still under discussion among clinicians. To deal with this situation, the Beckman Initiative for Macular Research Classification Committee proposed a new clinical classification scheme for AMD in 2013. The Beckman AMD classification system provides a simplified and unified guidance for broad clinical phenotypes by using a modified Delphi technique (Table 1) [41]. Based on the Beckman AMD classification system, the disease is classified into early-stage AMD, intermediate-stage AMD, and late-stage AMD (GA and neovascular AMD) [41]. Early-stage AMD is characterized by the presence of medium-sized drusen (>63 and ≤125 μm) without any impairment of visual function [41]. Intermediate-stage AMD is defined as the presence of large drusen (>125 μm) or/and abnormalities in the RPE [41]. Late-stage AMD (advanced AMD) is classified into two clinical forms: GA (dry or non-exudative AMD) and neovascular AMD (wet or exudative AMD) [41]. GA is defined by the irreversible loss of the RPE and photoreceptor cells, leading to a decrease in visual function [41]. Neovascular AMD is characterized by the invasion of newly fragile choroidal blood vessels growing from the choroid into the retina [41]. This process is known as choroidal neovascularization (CNV), which goes along with blood and fluid leakage, leading to detachment of the retina or RPE and rapid vision loss [41]. Figure 2 describes the clinical manifestation and pathology of AMD from early to late stage.
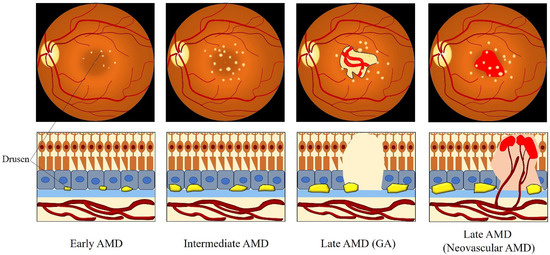
Figure 2. Clinical manifestation and pathology of AMD from the early to late stage. Medium-sized drusen found in early AMD. Intermediate AMD shows the presence of large drusen. Late AMD is classified into GA and neovascular AMD. GA is defined by the deterioration of the RPE, photoreceptor layer, and choroidal capillaries in the macula. The invasion of abnormal fragile choroidal blood vessels growing from the choroid into the retina in neovascular AMD, with blood and fluid leakage. Abbreviations: AMD: age-related macular degeneration; GA: geographic atrophy.
Table 1. The Beckman clinical classification of age-related macular degeneration (AMD) [41].
| Classification | Clinical Manifestation |
|---|---|
| No AMD | No drusen and no RPE abnormalities |
| Normal aging changes | Drusen ≤ 63 μm and no RPE abnormalities |
| Early AMD | Drusen > 63 μm and ≤125 μm and no RPE abnormalities |
| Intermediate AMD | Drusen > 125 μm and/or RPE abnormalities |
| Late AMD | GA and/or neovascular AMD |
Abbreviations: AMD: age-related macular degeneration; GA: geographic atrophy; RPE: retinal pigment epithelium.
4. Pathogenesis of AMD
4.1. Oxidative Stress and AMD
4.1.1. The Macula—An Ideal Environment for the Generation of ROS
It is well known that the retina is one of the highest oxygen-consuming tissues in the human body, utilizing even more oxygen per weight than the brain [42]. The local oxygen metabolic environment in the retina plays an essential role in keeping retinal homeostasis between the supply and consumption of retinal oxygen [42]. The retina continuously transforms light into vision, requiring a marked amount of energy and generating reactive oxygen species (ROS), such as the superoxide (O2•−), the hydroxyl radical (•OH), hydrogen peroxide (H2O2), and singlet oxygen (1O2) as normal metabolic byproducts [42]. Generally, ROS are produced during oxidative metabolism under physiological conditions and participate in normal cellular metabolism [43]. However, when the generation of ROS exceeds the capacity of the antioxidant systems, ROS disrupt the balance of redox homeostasis and cause oxidative stress [22].
Owing to the property of high oxygen-metabolism, retinal tissue generates significant amounts of ROS, which makes the retina susceptible to oxidative damage [22]. Evidence shows that the choroidal circulation can only supply the outer retina, while the inner retina is nourished by the retinal vasculature [44]. Importantly, the central 250–600 µm of the macula are devoid of retinal blood vessels and receive blood supply from the underlying choriocapillaris only [33]. Consequently, the RPE is exposed to high ambient oxygen partial pressures of 70–90 mm Hg, which provides an ideal environment for the generation of abundant exogenous ROS [45]. Moreover, the RPE constitutes the outer blood-retinal barrier (oBRB), maintains phagocytosis of around 30,000 photoreceptor outer segments, heat exchange, and vitamin A metabolism, which all produce high levels of ROS [46]. Furthermore, the macula is constantly exposed to light and absorbs light to optimize vision, which causes photo-oxidative stress as an additional source of exogenous oxidative stress [33][47]. An in vitro study showed that short-wavelength light induces ROS production in the mitochondria [48]. The characteristics of the unique sources of retinal ROS generation and high oxygen consumption suggest that oxidative damage is an essential factor in the mechanism of AMD development.
4.1.2. Animal Models of AMD
Experimental models of AMD have been developed in different species, such as mice, rats, rabbits and pigs to better understand the pathogenesis of the disease and to provide suitable preclinical models for drug intervention [49]. These animal models include natural and genetically engineered animal models [50]. A light-induced model is provided for AMD to imply the relationship between oxidative damage and AMD, since light is a natural risk factor involved in AMD, and a light-induced model is easy to produce by varying light intensity and duration [51][52][53][54]. Moreover, transgenic animal models have been studied in AMD over the last few years, such as the nuclear factor-erythroid 2-related factor 2 (Nrf2) knock-out (KO) mouse model, the peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) KO mouse model, the superoxide dismutase-2 (SOD2) KO mouse model and the peroxisome proliferator-activated receptor-β (PPARβ) KO mouse model [55][56][57]. These genetic animal models can develop various forms of AMD and enhance the understanding of the disease, offering great possibilities for gene therapeutic approaches.
4.1.3. The Generation of ROS Due to Light Exposure
It is well established that light exposure has the potential to cause detrimental effects in many organs and tissues, such as the skin, cornea, conjunctiva, lens, as well as the RPE and retina [58][59][60]. Large amounts of ROS are produced by exposure to ultraviolet light wavelengths from 100 nm to 400 nm and to blue light wavelengths from 400 nm to 500 nm [58][59][60]. It should be noted that the photoreceptors in the macula are directly exposed to light and are absorbing parts of the light spectrum through rhodopsin, a photoreceptor molecule in rods [61]. Since the cornea, anterior chamber, and crystalline lens effectively filter different parts of the ultraviolet spectrum, only a low portion (1% or less) of the ultraviolet band (315–400 nm) reaches the retina [62]. Some studies demonstrated that absorption of ultraviolet rays by the retina results in photochemical reactions via a type 1 mechanism (direct reactions involving proton or electron transfers) and a type 2 mechanism (reactions involving ROS) [60][62]. Likewise, blue light was shown to be capable of inducing substantial ROS formation in the retina and RPE. The generation of ROS during photooxidative stress may damage cellular components (lipids, proteins and deoxyribonucleic acid (DNA)) and, thereby, is responsible for a large part of cytotoxicity [63].
Based on an in vitro study on an organotypic culture system for mouse retinas by Roehlecke and Schumann, it has been proposed that the generation of ROS occurs directly in outer segments of photoreceptors by nicotinamide adenine dinucleotide phosphate oxidase (NOX) as well as by the mitochondria-like activity of the outer segments after visible blue light (405 nm) irradiation with an output power of 1 mW/cm2 [63]. In addition, the authors found that blue light rapidly induced ROS generation in photoreceptors of retinal explants after 0.5–1 h by increasing NOX activity (especially NOX2 and NOX4) and demonstrated that a cross-talk between NOX and mitochondria-like activity may stimulate NOX activation [63]. Another study conducted on the human RPE cell line, adult retinal pigment epithelial cell line-19 (ARPE-19), revealed that the mitochondrial electron transport chain is an important source of ROS and that mitochondria-derived ROS played a critical role in the death of cells exposed to short-wavelength blue light (425 ± 20 nm) [48]. Meanwhile, several pigments in the retina, such as rhodopsin, lipofuscin and melanin were shown to be involved in the process of inducing oxidative stress [64][65][66]. Grimm et al. reported that rhodopsin mediated blue-light-induced damage in the retina, which occurred after short time exposure to blue light but not to green light [64]. In the RPE, lipofuscin is derived primarily from phagocytosis of shed photoreceptor outer segments and is considered a heterogeneous waste material that accumulates with age in active postmitotic cells, such as those of the RPE [67][68]. Evidence also implicated that lipofuscin serves as a photoinducible generator of ROS and is an initiator of blue light damage in the RPE [31]. In an in vitro study in cultured RPE cells, Shamsi et al. demonstrated that lipofuscin is capable of inducing lipid peroxidation and reducing the efficacy of the lysosomal and antioxidant enzyme systems in RPE cells [69]. In an ARPE-19 cell culture model constructed by Sparrow et al., the lipofuscin fluorophore, N-retinyl-N-retinylidene ethanolamine (A2E), contributed to blue light-induced damage in RPE cells [65]. Furthermore, lipofuscin as a potent generator of ROS may exert deleterious effects to the retina by light exposure [70].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22031296
References
- Pennington, K.L.; DeAngelis, M.M. Epidemiology of age-related macular degeneration (amd): Associations with cardiovascular disease phenotypes and lipid factors. Eye Vis. 2016, 3, 34.
- Adelson, J.D.; Bourne, R.R.A. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to vision 2020: The right to sight: An analysis for the global burden of disease study. Lancet Glob. Health 2021, 9, e144–e160.
- Ke, K.M.; Chakravarthy, U. Economic cost of age-related macular degeneration. Drugs Aging 2006, 23, 217–225.
- Jonas, J.B.; Cheung, C.M.G. Updates on the epidemiology of age-related macular degeneration. Asia Pac. J. Ophthalmol. 2017, 6, 493–497.
- Wong, W.L.; Su, X. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116.
- Brown, M.M.; Brown, G.C. Age-related macular degeneration: Economic burden and value-based medicine analysis. Can. J. Ophthalmol. 2005, 40, 277–287.
- Donders, F.C. Beiträge zur pathologischen anatomie des auges. Arch. Für Ophthalmol. 1855, 1, 106–118.
- Nagel, A. Ueber chorioiditis areolaris und über krystalle im augenhintergrund. Areolar Choroiditis Cryst. Eye Fundus. Klin Monbl Augenheilk 1868, 4, 417–420.
- De Jong, P.T.V.M. A historical analysis of the quest for the origins of aging macula disorder, the tissues involved, and its terminology: Supplementary issue: Ophthalmic history. Ophthalmol. Eye Dis. 2016, 8.
- Haider, N.B.; Cruz, N.M. Pathobiology of the outer retina: Genetic and nongenetic causes of disease. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: San Diego, CA, USA, 2014; pp. 2084–2114.
- Prevention and Treatment of Age-related Macular Degeneration. Available online: (accessed on 14 November 2020).
- Lim, L.S.; Mitchell, P. Age-related macular degeneration. Lancet 2012, 379, 1728–1738.
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39.
- Bowes Rickman, C.; Farsiu, S. Dry age-related macular degeneration: Mechanisms, therapeutic targets, and imaging. Investig. Ophthalmol. Vis. Sci. 2013, 54, ORSF68–ORSF80.
- Chakravarthy, U.; Wong, T.Y. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 2010, 10, 31.
- Chakravarthy, U.; Augood, C. Cigarette smoking and age-related macular degeneration in the eureye study. Ophthalmology 2007, 114, 1157–1163.
- Cruickshanks, K.J.; Klein, R. Sunlight and the 5-year incidence of early age-related maculopathy: The beaver dam eye study. Arch. Ophthalmol. 2001, 119, 246–250.
- Tien, P.-T.; Lin, H.-J. Perfluorooctanoic acid in indoor particulate matter triggers oxidative stress and inflammation in corneal and retinal cells. Sci. Rep. 2020, 10, 15702.
- Bellezza, I. Oxidative stress in age-related macular degeneration: Nrf2 as therapeutic target. Front. Pharmacol. 2018, 9, 1280.
- Lipecz, A.; Miller, L. Microvascular contributions to age-related macular degeneration (amd): From mechanisms of choriocapillaris aging to novel interventions. GeroScience 2019, 41, 813–845.
- Rakoczy, P.E.; Zhang, D. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am. J. Pathol. 2002, 161, 1515–1524.
- Beatty, S.; Koh, H.-H. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv. Ophthalmol. 2000, 45, 115–134.
- Archer, D.; Gardiner, T.J.A.j.o.o. Electron microscopic features of experimental choroidal neovascularization. Am. J. Ophthalmol. 1981, 91, 433–457.
- Stone, E.M.; Braun, T.A. Missense variations in the fibulin 5 gene and age-related macular degeneration. N. Engl. J. Med. 2004, 351, 346–353.
- Chou, M.Y.; Hartvigsen, K. Oxidation-specific epitopes are important targets of innate immunity. J. Intern. Med. 2008, 263, 479–488.
- The Anatomy of the Macula. Available online: (accessed on 15 November 2020).
- Margolis, R.; Spaide, R.F. A pilot study of enhanced depth imaging optical coherence tomography of the choroid in normal eyes. Am. J. Ophthalmol. 2009, 147, 811–815.
- The Macula. Available online: (accessed on 27 January 2021).
- Tschulakow, A.V.; Oltrup, T. The anatomy of the foveola reinvestigated. PeerJ 2018, 6, e4482.
- Curcio, C.A.; Sloan, K.R. Human photoreceptor topography. J. Comp. Neurol. 1990, 292, 497–523.
- Sparrow, J.R.; Hicks, D. The retinal pigment epithelium in health and disease. Curr. Mol. Med. 2010, 10, 802–823.
- Hughes, B.A.; Gallemore, R.P.; Miller, S.S. The Retinal Pigment Epithelium: Current Aspects of Function and Disease; Marmor, M.F., Wolfensberger, T.J., Eds.; Oxford University Press: New York, NY, USA, 1998; pp. 103–134.
- Handa, J.T. How does the macula protect itself from oxidative stress? Mol. Asp. Med. 2012, 33, 418–435.
- Fryczkowski, A.W. Anatomical and functional choroidal lobuli. Int. Ophthalmol. 1994, 18, 131–141.
- Sivaprasad, S.; Bailey, T.A. Bruch’s membrane and the vascular intima: Is there a common basis for age-related changes and disease? Clin. Exp. Ophthalmol. 2005, 33, 518–523.
- Okubo, A.; Rosa, R.H., Jr. The relationships of age changes in retinal pigment epithelium and bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 1999, 40, 443–449.
- Davis, M.D.; Gangnon, R.E. The age-related eye disease study severity scale for age-related macular degeneration: Areds report no. 17. Arch. Ophthalmol. 2005, 123, 1484–1498.
- Age-Related Eye Disease Study Research Group. Risk factors associated with age-related macular degeneration. A case-control study in the age-related eye disease study: Age-related eye disease study report number 3. Ophthalmology 2000, 107, 2224–2232.
- Mitchell, P.; Foran, S. Age-related eye disease study severity scale and simplified severity scale for age-related macular degeneration. Arch. Ophthalmol. 2005, 123, 1598–1599.
- Ferris, F.L.; Davis, M.D. A simplified severity scale for age-related macular degeneration: Areds report no. 18. Arch. Ophthalmol. 2005, 123, 1570–1574.
- Ferris, F.L.; Wilkinson, C.P. Clinical classification of age-related macular degeneration. Ophthalmology 2013, 120, 844–851.
- Yu, D.-Y.; Cringle, S.J. Retinal degeneration and local oxygen metabolism. Exp. Eye Res. 2005, 80, 745–751.
- Ray, P.D.; Huang, B.-W. Reactive oxygen species (ros) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990.
- Sun, Y.; Smith, L.E.H. Retinal vasculature in development and diseases. Annu. Rev. Vis. Sci. 2018, 4, 101–122.
- Winkler, B.S.; Boulton, M.E. Oxidative damage and age-related macular degeneration. Mol. Vis. 1999, 5, 32.
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881.
- Miceli, M.V.; Liles, M.R. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp. Cell. Res. 1994, 214, 242–249.
- King, A.; Gottlieb, E. Mitochondria-derived reactive oxygen species mediate blue light-induced death of retinal pigment epithelial cells. Photochem. Photobiol. 2004, 79, 470–475.
- Pennesi, M.E.; Neuringer, M. Animal models of age-related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509.
- Chader, G.J. Animal models in research on retinal degenerations: Past progress and future hope. Vis. Res. 2002, 42, 393–399.
- Duncan, J.L.; LaVail, M.M. Intense cyclic light-induced retinal degeneration in rats. Arch. Ophthalmol. 2010, 128, 244–245.
- Wielgus, A.; Collier, R. Blue light induced a2e oxidation in rat eyes–experimental animal model of dry amd. Photochem. Photobiol. Sci. 2010, 9, 1505–1512.
- Wenzel, A.; Grimm, C. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog. Retin. Eye Res. 2005, 24, 275–306.
- Abokyi, S.; To, C.-H. Central role of oxidative stress in age-related macular degeneration: Evidence from a review of the molecular mechanisms and animal models. Oxid. Med. Cell. Longev. 2020, 2020, 7901270.
- Felszeghy, S.; Viiri, J. Loss of nrf-2 and pgc-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12.
- Justilien, V.; Pang, J.-J. Sod2 knockdown mouse model of early amd. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4407–4420.
- Choudhary, M.; Ding, J.-D. Pparβ/δ selectively regulates phenotypic features of age-related macular degeneration. Aging 2016, 8, 1952–1978.
- Nakashima, Y.; Ohta, S. Blue light-induced oxidative stress in live skin. Free Radic. Biol. Med. 2017, 108, 300–310.
- Marek, V.; Mélik-Parsadaniantz, S. Blue light phototoxicity toward human corneal and conjunctival epithelial cells in basal and hyperosmolar conditions. Free Radic. Biol. Med. 2018, 126, 27–40.
- Chalam, K.V.; Khetpal, V. A review: Role of ultraviolet radiation in age-related macular degeneration. Eye Contact Lens 2011, 37, 225–232.
- Funk, R.H.; Schumann, U. Blue light induced retinal oxidative stress: Implications for macular degeneration. World J. Ophthalmol. 2014, 4, 29–34.
- Glickman, R.D. Ultraviolet phototoxicity to the retina. Eye Contact Lens 2011, 37, 196–205.
- Roehlecke, C.; Schumann, U. Stress reaction in outer segments of photoreceptors after blue light irradiation. PLoS ONE 2013, 8, e71570.
- Grimm, C.; Wenzel, A. Rhodopsin-mediated blue-light damage to the rat retina: Effect of photoreversal of bleaching. Investig. Ophthalmol. Vis. Sci. 2001, 42, 497–505.
- Sparrow, J.R.; Nakanishi, K. The lipofuscin fluorophore a2e mediates blue light–induced damage to retinal pigmented epithelial cells. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1981–1989.
- Dontsov, A.E.; Glickman, R.D. Retinal pigment epithelium pigment granules stimulate the photo-oxidation of unsaturated fatty acids. Free Radic. Biol. Med. 1999, 26, 1436–1446.
- Sparrow, J.R.; Boulton, M. Rpe lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606.
- Brunk, U.T.; Terman, A.J.F.R.B. Lipofuscin: Mechanisms of age-related accumulation and influence on cell function. Free Radic. Biol. Med. 2002, 33, 611–619.
- Shamsi, F.A.; Boulton, M. Inhibition of rpe lysosomal and antioxidant activity by the age pigment lipofuscin. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3041–3046.
- Ng, K.-P.; Gugiu, B. Retinal pigment epithelium lipofuscin proteomics. Mol. Cell. Proteom. 2008, 7, 1397–1405.