Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Protein biosynthesis is a vital process for all kingdoms of life. The ribosome is the massive ribonucleoprotein machinery that reads the genetic code, in the form of messenger RNA (mRNA), to produce proteins. The mechanism of translation is tightly regulated to ensure that cell growth is well sustained. In bacteria, the ribosome is a major target of inhibitors, as demonstrated by the high number of small molecules identified to bind to it. In eukaryotes, the design of ribosome inhibitors may be used as a therapy to treat cancer cells, which exhibit higher proliferation rates compared to healthy ones.
- ribosome
- protein synthesis inhibition
- anticancer drugs
- X-ray crystallography
- cryo-EM
- drug development
1. Introduction
The ribosome is the universal ribonucleoprotein machinery that translates the genetic message stored in the mRNA into proteins. Its function is highly conserved and tightly regulated in all domains of life. The ribosome is composed of two subunits, one large ribosomal subunit (LSU; 50S in bacteria and 60S in eukaryotes, depending on their sedimentation coefficient (Svedberg unit)) and a small ribosomal subunit (SSU; 30S in bacteria and 40S in eukaryotes). However, its composition varies remarkably between prokaryotes and eukaryotes. Ribosomes in bacteria are constituted by three types of ribosomal RNA (rRNA) and 54 individual ribosomal proteins (RPs), reaching a molecular weight of approximately 2.5 MDa. In eukaryotes, instead, ribosomes exist as much more complex machineries. In the model organism Saccharomyces cerevisiae, for instance, the ribosome is made of four types of rRNA and 79 individual RPs, reaching a molecular weight of 3.3 MDa. Further compositional differences exist between higher and lower eukaryotes, with mammalian ribosomes containing an additional RP and, more importantly, 1 MDa of rRNA extensions called expansions segments (ESs) [1,2].
The key role played by the ribosome within the cell has made it one of the favourite targets of antibiotics in bacteria (reviewed in [14,15,16,17,18]). These small molecules, synthesised by fungi or soil bacteria as a self-defence mechanism, interact within the functional sites of the ribosome, thus hindering translation. Extensive studies in the past (for more detail see reviews above), but also more recent ones [19,20,21,22], have shed light on the functional implications of protein synthesis inhibition triggered by the binding of antibiotics and on how bacteria can develop resistance. Different classes of antibiotics have been shown to cluster in the Aminoacyl (A), Peptidyl (P) and Exit (E) binding sites within the 50S and 30S subunits [14,17]. Additionally, antimicrobial compounds have been identified to tether to the SSU either to prevent the binding or, very surprisingly, to stall the movement of mRNA during elongation [15]. Structural works focused on unravelling the binding modes of these molecules within the ribosome have been, and yet are, of paramount importance for understanding the molecular basis of inhibition in bacteria and guiding drug design strategies to develop novel antibiotics able to elude resistance mechanisms [23].
Intriguingly, but not totally surprisingly, the eukaryotic ribosome is also the target of naturally derived small molecule inhibitors (Figure 1 and Figure 2, Supplementary Table S1 and Supplementary Session S1). Several compounds are known to hinder translation at different steps (Figure 1), as demonstrated by extensive functional works [24,25,26,27,28,29,30] (and reviewed in [31,32,33]). But until recently, no atomic-resolution information on the mode of action of ribosome inhibitors in eukaryotes was available. The later advances in crystal structure determination of 80S ribosomes from the model organism S. cerevisiae allowed applying X-ray crystallography to solve the first high-resolution structures of inhibitors in complex with the eukaryotic ribosome. In this first study, the molecular details of binding of 12 eukaryotic-specific and 4 broad-spectrum inhibitors in complex with the yeast 80S ribosome was unveiled [34]. All compounds unambiguously fit within the vacant 80S ribosome and were revealed to cluster at ribosomal functional sites, similarly to bacteria (Figure 1 and Figure 2, [34].
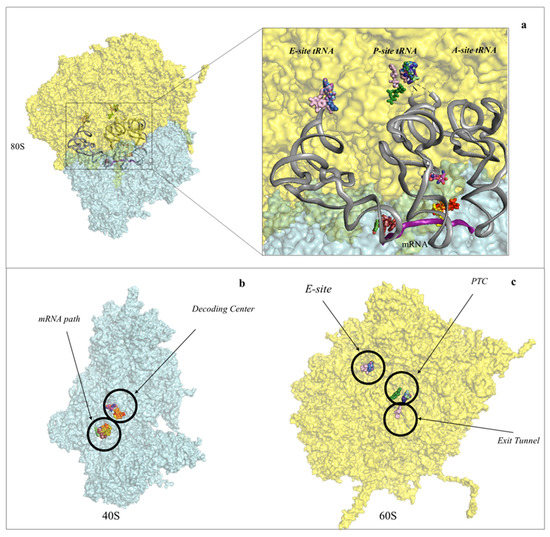
Figure 2. Protein synthesis inhibitors cluster to ribosomal functional sites. (a) Left-hand side, overview of the eukaryotic 80S ribosome, shown as surface representation. LSU is coloured in cyan, SSU in pale yellow. The three tRNAs and mRNA were modelled in silico after superposition of the Thermus thermophilus elongating complex (PDB: 4V5D) to the yeast vacant 80S model (PDB: 4V88). Right-hand side, zoom-in of the regions where inhibitors bind within the ribosome. For clarity the functional sites are labelled. PTC: peptidyl-transferase centre; PET: peptide exit tunnel; DC: decoding centre; (b) binding of small molecule inhibitors to the eukaryotic SSU. Drugs are represented as spheres, SSU as surface. The functional sites targeted are displayed; (c) binding of small molecule inhibitors to the eukaryotic LSU. Drugs are represented as spheres, SSU as surface.
In addition, there is increasing evidence that rRNA modifications and pervasive sequence variation in rRNA can be related to differential expression in human cells and tissues to originate “specialised” ribosomes, suggesting also a possible link with cancer onset [41,42,43,44]. Different patterns of rRNA modifications expand further the complexity of designing more potent and highly specific anticancer drugs. Visualising these “specialised” ribosomes will provide new understanding at functional level but, more importantly, will serve to gather a much more refined structural interpretation of the ribosomal druggable sites.
2. Inhibitors Targeting the Large Ribosomal Subunit
The peptidyl transferase centre is the region within the LSU where peptide bond formation occurs. It requires the aminoacyl-tRNA and peptidyl-tRNA to be properly accommodated in the A- and P-sites, respectively; correct positioning of the two tRNAs promotes nucleophilic attack of the amino-terminal of the amino acid carried by the A-site tRNA towards the carbonyl group of the elongating nascent chain [45,46]. Upon peptide bond formation, the new polypeptide chain elongates and finally protrudes out from the peptide exit tunnel (PET), where it encounters dedicated chaperones that promote its correct folding [47]. Once discharged, the deacylated tRNA moves further towards the E-site and, through concerted movements of the L1-stalk, is allowed to exit the ribosome.
Structural studies in bacteria and eukaryotes have confirmed that one of the best strategies to halt translation is by interfering with all these steps during elongation (Figure 1 and Figure 2 and reviewed in [14,17,18]). The PTC is one of the major binding sites for inhibitors targeting the LSU, especially for members of the alkaloid and mycotoxin families (Figure 3) [14,34,48,49]. It was known that the PET is targeted by a large family of compounds in bacteria, the macrolides; however, recent studies have demonstrated that this region can also be targeted in eukaryotes (Figure 4) [16,20,50]. Finally, the achievement of high-resolution structures of the yeast and human ribosomes allowed unveiling the binding modes of inhibitors targeting the E-site of the LSU, an exclusive eukaryotic-specific feature (Figure 5) [34,51,52].
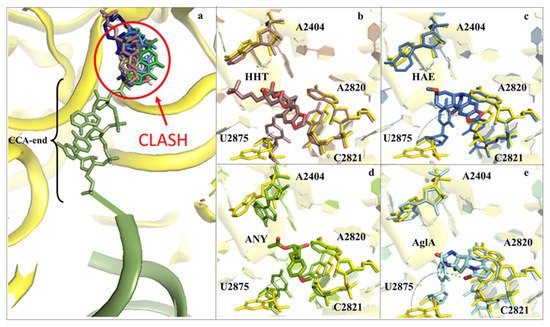
Figure 3. Inhibitors binding to the A-site cleft impede the binding of the aa-tRNA during elongation. (a) Overview of the inhibitors that bind to the A-site cleft. All compounds would sterically clash with the incoming charged acylated A-site tRNA, thus impairing accommodation of the following amino acid within the pocket and therefore halting the elongation phase of translation. Anisomycin (ANI) in lemon (PDB: 4U3M), agelastatin A (AglA) in cyan (PDB: 5MEI), deoxyvalenol in fuchsia (PDB: 4U53), haemanthamine (HAE) in blue (PDB: 5ON6), homoharringtonine (HHT) in brown (PDB: 4U4Q), lycorine (LYC) in light blue (PDB: 4U4U), nagilactone C in aquamarine (PDB: 4U52), narciclasine (NAR) in green forest (PDB: 4U51), T2-toxin in purple (PDB: 4U6F) and verrucarin A (PDB: 4U50) in blue navy; (b) zoom-in and details of interaction for HHT within the A-site cleft. Proximal rRNA residues are shown as sticks; (c) zoom-in and details of interaction for HAE within the A-site cleft; (d) zoom-in and details of interaction for ANI within the A-site cleft; (e) zoom-in and details of interaction for AglA within the A-site cleft. The rRNA residues that adopt different conformations upon binding of the inhibitors, A2404 and U2875, are shown for clarity. The peculiar halogen–π interaction established between the pyrimidine ring of U2875 and the bromine atom of AglA is also highlighted.
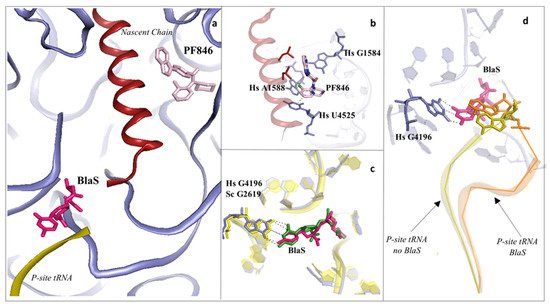
Figure 4. Inhibitors binding to the PTC impair elongation of the nascent chain and can be sequence specific. (a) Overview of the PTC and peptide exit tunnel (PET) of the human ribosome. Blasticidin S (BlaS) in green and hot pink (PDB: 4U56 Sc/7NWI Hs) and PF846 in light pink (PDB: 6XA1) bind within the PTC, where the P-site tRNA and nascent chain, respectively, interact with the ribosome; (b) PF846 binds to the PET and contacts residues A1588 and U4525 by establishing two different types of interactions, halogen–π and π–π stacking, respectively, each mediated by the chloride atom; (c) structural superimposition of human and yeast ribosomes solved in complex with BlaS. The conserved residue G4196 (G2619 yeast numbering) establishes a similar hydrogen bonding network with the compound in both organisms; (d) superimposition of translating human ribosome complexes in presence (PDB: 7NWI) and absence of BlaS (PDB: 6XA1). Detail of the displacement of the CCA-end of the P-site tRNA upon binding of the drug, which further promotes an additional π–π stacking with A76, likely preventing the correct accommodation of the tRNA into the PTC. P-site tRNA in absence of BlaS is coloured in olive, while the P-site tRNA in presence of BlaS in orange. Hs: H. sapiens.
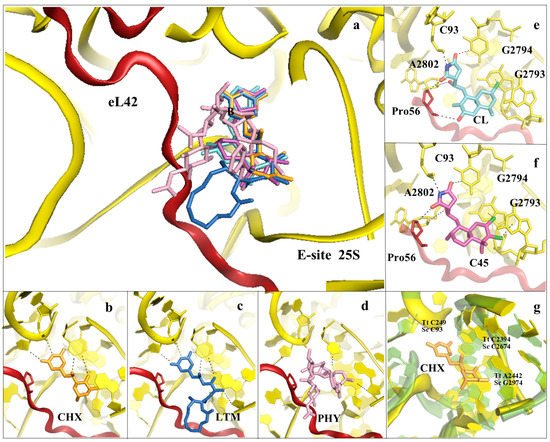
Figure 5. Inhibitors binding to the E-site of the LSU impair the translocation process. (a) Overview of the inhibitors targeting specifically the eukaryotic E-site. Cycloheximide (CHX) (PDB: 4U43) in orange, lactimidomycin (LTM) in blue (PDB: 4U4R), phyllantoside (PHY) in pink (PDB: 4U4Z), chlorolissoclimide (CL) in light cyan (PDB: 5TBW) and the synthetic analogue C45 in magenta (PDB: 6HHQ); (b) zoom-in and details of interaction for CHX within the E-site binding pocket; (c) zoom-in and details of interaction for LTM within the E-site binding pocket; (d) zoom-in and details of interaction for PHY within the E-site binding pocket. PHY, in contrast to CHX and LTM, is reported to interact directly with the eukaryotic-specific ribosomal protein eL42. eL42 is coloured in red for clarity; (e) zoom-in of CL interaction network within the E-site, with details of the hydrogen bonding and the peculiar halogen–π interaction that lissoclimides establish with rRNA residues. Additionally, CL interacts with the backbone of eL42; (f) same view for C45, with highlighted the second halogen–π interaction that the drug establishes with the rRNA. This additional bond might compensate for the lack of the hydrogen bond with eL42, as it occurs with CL. In both images, the type of bonding established between the drug and the ribosome is marked; (g) structural superimposition of the S. cerevisiae LSU (coloured in yellow) with the LSU of T. thermophilus (coloured in light green) (PDB: 4V5D) (rmsd 4.5 Å). Numbers of residues for the two organisms are shown (Sc: S. cerevisiae, Tt: T. thermophilus).
Very interestingly, specific translation elongation inhibitors, such as anisomycin (ANI), lactimidomycin (LTM) and deoxynivalenol, trigger significant activation of the JNK/p38 MAPK signalling pathway, leading to rRNA cleavage and cell death (reviewed in [31]). Importantly, a recent work has demonstrated that this process, called a ribotoxic stress response (RSR), is initiated by the formation of a few colliding ribosomes at intermediate concentrations of inhibitor (in the specific case, ANI) [53] (and reviewed in [31]).
3. Inhibitors Targeting the 40S Small Ribosomal Subunit
The SSU is responsible for scanning the mRNA to initiate translation. Additionally, it ensures the accurate decoding of the genetic code during the elongation phase. There are two main regions where inhibitors have been identified to bind to: the mRNA path and the decoding centre (DC) (Figure 2, Figure 6 and Figure 7).
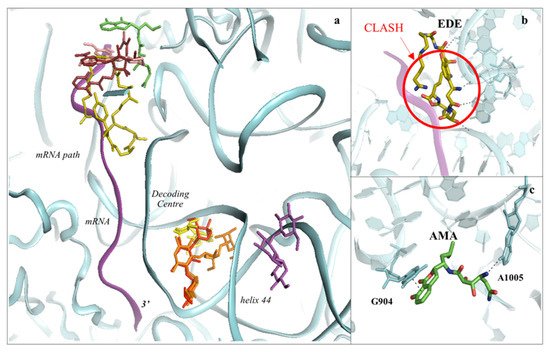
Figure 6. The SSU can be targeted by protein synthesis inhibitors at two main regions. (a) Overview of the binding sites for small molecules binding to the 40S subunit. Amicoumacin A (AMA) in green (PDB: 5I4L), cryptopleurine in salmon (PDB: 4U55), edeine (EDE) in olive green (PDB: 4U4N) and pactamycin (PAC) in ruby (PDB: 4U4Y). mRNA is also shown as a ribbon and coloured in purple. Inhibitors have been shown to bind with high specificity to the mRNA path and the decoding centre (DC), thus impairing tRNA–mRNA translocation and altering the translation fidelity process; (b) zoom-in and details of interaction for EDE within the mRNA path in correspondence with where the E-site tRNA should interact. The clash with mRNA (shown in red) would prevent the formation of the preinitiation complex; (c) zoom-in and details of interaction for AMA within the mRNA path. AMA forms a π–π stacking interaction with the conserved rRNA residue G904 while hydrogen bonding with the conserved A1005 (shown as dashed lines).
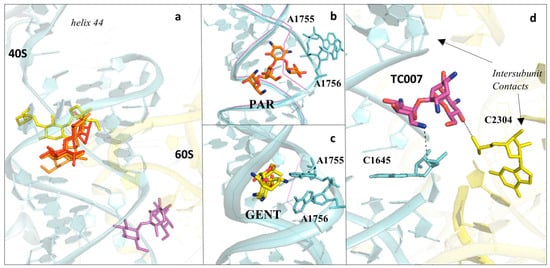
Figure 7. Aminoglycosides bind preferentially to the DC of the eukaryotic ribosome. (a) Overview of aminoglycosides binding in correspondence of helix 44 (h44) of the eukaryotic SSU. Paromomycin (PAR) in orange (PDB: 5NDV), geneticin (G418) in red (PDB: 5NDG), gentamycin (GENT) in yellow (PDB: 5OBM), TC007 in violet (PDB: 5NDJ); (b) PAR binds within the minor groove of h44 and promotes “flip-out” of the two conserved adenines (A1755/A1756) towards the mRNA channel; (c) GENT binds to h44, but docks in a different way compared to PAR, and induces only partial extrusion of the decoding residues. Vacant ribosome (PDB: 4V88) aligned and shown as a purple line for evidencing the conformational change of the minor groove of h44 upon drug binding; (d) zoom-in and details of interaction for the aminoglycoside TC007. TC007 directly contacts both LSU and SSU, establishing hydrogen bonds with rRNA residues of the 25S and 18S at a specific intersubunit bridge (B3).
4. Conclusions and Future Directions
The recent advances in the field of cryo-EM have made it possible to achieve atomic-resolution reconstructions, as recently proven [91,92]. This is currently restricted to few very well-established examples; however, continuous development in EM hardware and single-particle processing software, coupled to higher knowledge of optimisation strategies for grid freezing conditions, might enhance the feasibility of achieving sub-2 Å overall resolution reconstructions, as shown by the work on the E. coli 70S ribosome [93]. At such a level of detail, although not attainable for solvent exposed regions and flexible domains, the solvation shells within inhibitors’ binding pockets become interpretable and can further guide drug development through optimisation of chemical functionalisation of existing compounds. Additionally, the preparation of cryo-EM grids is a rapid process and permits working with concentrations of drugs in the low micromolar range. Crystallisation, conversely, is a slow process (ribosome crystals take approximately 3–4 weeks to reach their final size), and unstable compounds can degrade during this time. To overcome this, grown crystals can be soaked into solutions containing the drug of interest. However, because of the highly viscous components at this stage, which could reduce diffusion of small molecules into the ribosome, high concentrations must be used (in some cases up to the millimolar range), thus requiring significant amounts of compounds. On the other hand, despite the hurdle of producing high-quality ribosome crystals, the great benefit is still the relatively medium–high throughput at which compounds can be screened, with results achievable in the order of 36–48 h post-data collection at a synchrotron source. Electron density maps in the range of 2.9–3.5 Å resolution can provide compelling details about the region of binding and the interaction pattern that a drug establishes within the ribosome and could serve to narrow down the search for the optimal inhibitor.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13174392
This entry is offline, you can click here to edit this entry!