1. Introduction
The human gastrointestinal tract (GIT) harbors a great diversity of microorganisms known as the gut microbiota [
1,
2]. The gut microbiota forms complex communities that coexist in an intimate relationship with the host, providing great benefits such as metabolic products and favoring the development of the immune system [
3,
4]. These gut microbial communities are present as planktonic cells or biofilm communities [
5].
2. Health and Disease: Non-Invasive versus Invasive Gut Microbial Biofilms
In a healthy gut, a beneficial microbial biofilm formed by a complex ecological community will interact with the mucus layer and epithelium without invading the epithelia layer. This allows essential functions such as microbiota stability and resilience, which contribute to gut homeostasis and protect against infections [
4,
5]. Commensal biofilms offer a protective barrier against the proliferation and colonization of enteric pathogens, as well as of opportunistic pathobionts [
41]. The resistance mechanisms offered by commensal communities against enteropathogens include the use of bacteriocins and short-chain fatty acids production, which inhibits the growth of pathogens and pathobionts [
42,
43,
44]. Furthermore, commensal bacteria facilitate the host barrier function by thickening the mucus layer, inducing the expression of antimicrobial molecules and regulating the secretion of IgA [
45,
46,
47,
48]. Moreover, commensal microorganisms stimulate conversion of pro-IL-1β into active IL-1β [
49] and induce the development of Th17 cells in the intestine, allowing protection against pathogens [
50] (
Figure 1).
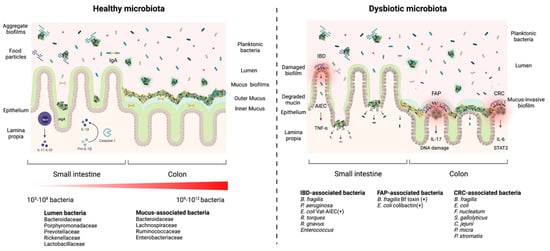
Figure 1. Healthy microbiota biofilms versus a dysbiotic microbiota biofilms. In a healthy microbiota (left panel), the microbial density and diversity increase from the stomach to the colon. In the small intestine, biofilms are discontinuous and loose aggregates, while in the large intestine, biofilms are dense, continuous and attached to a uniform mucus layer (attached biofilms). The biofilms in the gut lumen are loosely attached to food particles or encapsulated in mucin (aggregate biofilms). Commensal biofilms facilitate the host barrier function by thickening the mucus layer, regulating the secretion of IgA, stimulating conversion of pro-IL-1β into active IL-1β and inducing the development of Th17 cells. A dysbiotic microbiota (right panel) presents (1) damaged mucus-biofilm exposing epithelium cells to luminal content or (2) invasive biofilms where bacteria come directly into contact with the epithelium. Both scenarios expose the intestinal epithelium to pathogens and pathobionts which can trigger an infection. Invasive polymicrobial biofilms could trigger cellular inflammation, abnormal cellular proliferation, increased epithelial permeability (activation of IL-6 and Stat3) in patients with colorectal cancer (CRC), increased IL-17 production and DNA damage in patients with familial adenomatous polyposis (FAP), and inflammatory bowel disease (IBD). Patients’ Adherent-invasive E. coli (AIEC) colonize the intestinal mucosa and stimulate the secretion of TNF-α and mucin degradation.
On the other hand, when dysbiosis occurs, the physiological conditions in the gut are altered, which affects the organisation of the mucosal biofilm. These changes can result in two possible scenarios: (1) the mucosal biofilm is damaged and forms aggregates of different sizes which leads to the exposure of epithelial cells to luminal content; or (2) an invasive biofilm is formed, bacteria colonize the inner sterile mucus layer and potentially come directly into contact with the epithelium (
Figure 1). Both scenarios expose the intestinal epithelium to pathogens and pathobionts which can trigger an infection [
5]. For example, changes in diet, such as fiber deficiency, promote the expansion of colonic mucus-degrading bacteria in mice, leading to the erosion of the colonic mucus barrier and facilitating the access to epithelial cells for enteric pathogens that cause colitis in mice such as
Citrobacter rodentium [
51], a surrogate pathogen for enterohemorrhagic
E. coli (EHEC) and enteropathogenic
E. coli (EPEC) [
52].
Dysbiosis can also lead to invasive polymicrobial biofilms that induce cellular inflammation and abnormal cellular proliferation [
19]. Invasive biofilms are associated directly with tumors. A signature of invasive biofilms is the reduction of E-cadherin on the surface of colonic epithelial cells and the high activation of IL-6 and Stat3, which increase epithelial permeability and tissue inflammation [
19] (
Figure 1).
H. pylori is able to form biofilms in patients with peptic ulcer disease [
18].
H. pylori forms biofilm-like microcolonies deep in the stomach glands and interacts directly with gastric progenitor and stem cells in tissues from mice and humans. These gland-associated bacteria accelerate stem cell proliferation and up-regulate the expression of stem cell–related genes, leading to glandular hyperplasia [
53].
Bacterial biofilms present in the colon may also alter the host tissue microenvironment and induce metabolic changes in patients with colon cancer, as evident in metabolomic studies demonstrating changes in polyamine metabolite, including an upregulation of
N1,
N12-diacetylspermine. Increased polyamine concentrations are correlated with eukaryotic proliferation, potentially affecting cancer growth, development and progression [
54].
Furthermore, invasive polymicrobial biofilms associated with diseases are composed of specific bacterial species or groups. For example, invasive biofilms associated with the colonic mucosa of familial adenomatous polyposis (FAP) patients, an inherited disorder characterized by cancer of the large intestine, were predominately composed of
Escherichia coli and
Bacteroides fragilis. These bacteria can secrete oncotoxins named colibactin (ClbB) and
B. fragilis toxin (BFT), respectively, and these toxins were enriched in FAP patients. Furthermore, mice co-colonized with oncotoxin-producing strains had an increase in IL-17 production in the colon and increased DNA damage in colonic epithelial cells leading to faster onset of tumor [
55] creation. Specifically, the BFT toxin triggers a pro-carcinogenic multi-step inflammatory cascade that increases the production of genotoxic oxygen radicals in colonic epithelial cells [
56] (
Figure 1).
Patients with colorectal cancer (CRC) have a higher number of
Fusobacterium nucleatum and
Streptococcus gallolyticus that surround the carcinoma or the adenoma tissues [
57,
58]. Both bacteria possess virulence factors that stimulate inflammatory and oncogenic responses [
59]. Other bacteria that have been found in invasive biofilms in CRC patients are
Campylobacter jejuni, Parvimonas micra, and
Peptostreptococcus stomatis [
60,
61] (
Figure 1).
Similarly, invasive biofilms are also associated with inflammatory bowel disease (IBD) such as Crohn’s disease (CD) and ulcerative colitis (UC) [
16,
17]. In patients with IBD,
B. fragilis is responsible for more than 60% of the biofilm mass [
62]. Another study found a high proportion of pro-inflammatory bacteria on the colonic mucosa of a young patient with ulcerative colitis such as Enterobacteriaceae,
B. fragilis and
P. aeruginosa [
63]. Adherent-invasive
E. coli (AIEC) were isolated from ileal biopsies of 36.4% of patients with CD. AIEC colonize the intestinal mucosa, survive and then replicate in epithelial cells and macrophages, which stimulate the secretion of large amounts of TNF-α [
64] (
Figure 1). Interestingly, AIEC possess a protease called Vat-AIEC that favors mucosa colonization by degrading mucins and decreasing mucus viscosity [
65]. Also, an increased prevalence of mucolytic bacterial species such as
Ruminococcus gnavus and
Ruminococcus torques were found in CD and UC patients [
66]. Furthermore,
Enterococcus virulence factors were detected in children with IBD, and biofilm production was more frequent among
Enterococcus strains isolated from children with IBD than in control strains [
67] (
Figure 1).
Overall, certain intestinal pathologies create an ideal environment which foster enrichment of specific bacterial groups. Bacteria associated with disease will form low diversity biofilm communities that exacerbate underlying conditions whereas bacteria associated with health will form a highly diverse biofilm community that strengthens the natural defenses of the gut epithelium [
5]. Development and function of these biofilm communities will be influenced by host factors, host-microbe interactions, and microbe-microbe interactions [
5].
3. Diversity of Interactions and Phenotypes in the Gut Biofilm Communities
Interactions in mixed-species biofilm communities of the gut can be neutral, positive or negative. Positive interactions are characterized by cooperation, commensalism and cross-feeding, whereas negative interactions are characterized by competition, exploitation and interference [
68]. Cooperation involves one species that increases the fitness of another. Cooperation is not always reciprocal; however, if the interaction has a cost for one partner, an indirect benefit should be received for the interaction to be stable [
69]. Competition is an indirect interaction between two species competing for a common resource. For example,
Salmonella enterica induces inflammatory host responses that change the microbiota composition and suppress the microbiota’s growth [
70]. In the case of exploitation, one species gains a fitness benefit at the cost to another, and this is also known as predation or parasitism [
68]. Interference is a direct interaction where one species affects the fitness of another [
68]. Interference includes the use of bacteriocins [
71], type V, type VI and type VII secretion systems [
72,
73,
74]. Overall, different types of interactions are occurring in biofilms and these will shape the properties and the special arrangement of biofilm communities.
4. Diverse Gut and Microbiota-Derived Signals Induce Biofilm Formation in Commensal Bacteria and Enteropathogens
The transition from a planktonic state to sessile growth is regulated by multiple steps and regulation cascades, and includes QS-dependent genes, the type IV pili (T4P), and the flagellum [
104,
105,
106]. Biofilm formation is also guided by several environmental signals, which include mechanical signals, nutritional and metabolic cues, inorganic molecules, osmolarity, the presence of antimicrobial molecules, quorum-sensing derived signals, and host-derived signals [
107].
Bacteria can initiate the transition from a planktonic state to biofilm in vivo to improve their survival against harmful conditions present in the host, to exploit a nutrient rich area that facilitates colonization, or to use the cooperative benefits of multicellular structures [
108]. Biofilm formation can be controlled by stress response regulators that are activated by different stresses present in the host such as nutrient limitation, iron deprivation, sub-inhibitory concentrations of antibiotics, and osmotic stress [
109,
110,
111]. Specific environmental conditions such as calcium concentration can increase the second messenger c-di-GMP concentrations that could trigger biofilm formation [
112]. In some cases, biofilm formation is dependent on the nutritional conditions that will trigger metabolic adaptation and thus stimulate biofilm formation [
106]. In the next section, we will focus on host-derived signals that induce biofilm formation in gut commensal bacteria and enteropathogens.
4.1. Host-Derived Factors and Biofilm Formation
Bile salts present in the intestinal tract of the host can induce biofilm formation in several enteropathogens and improve their survival against the toxic effects of bile [
113]. Bile salts promote biofilm formation in
V. cholerae by increasing the intracellular levels of c-di-GMP, which are caused by an increase in c-di-GMP synthesis by 3 diguanylate cyclases (DGCs) and decreased expression of one phosphodiesterase (PDE) [
114]. The enteropathogen
Shigella flexnerii also forms biofilm in response to the presence of deoxycholate (DOC), and this is mediated by the secreted protein IcsA, which is involved in cell-cell contacts and aggregative growth [
115]. Similarly, vancomycin-resistant
Enterococcus (VRE) is able to form biofilms in the presence of physiological concentrations of bile acids, which facilitates colonization and persistence. In VRE, the ability to form biofilms in response to bile salts is controlled by the histine kinase YycG/Walk of the WalRK two component system and the response regulator LiaR of the three-component regulatory system LiaFSR [
116]. Likewise,
B. fragilis treatment with bile salts increased bacterial co-aggregation, adhesion to intestinal epithelial cells and biofilm formation [
117]. Exposure to bile salts induced morphological and transcriptional changes in
B. fragilis, including overproduction of fimbria-like appendages and outer membrane vesicles, and increased expression of genes encoding RND-type efflux pumps and the major outer membrane protein, OmpA [
117].
Additionally,
Acinetobater baumannii,
Cronobacter malonaticus, and
Bifidobacterium formed more biofilms when exposed to bile salts [
118,
119,
120]. In
Bifidobacterium breve, bile-salt-induced biofilm formation involved QS, EPS production and eDNA release, and increased its viability when exposed to porcine bile salts [
118]. In
A. baumannii, exposure to bile salts increased expression of virulence factors associated with surface motility, biofilm, and type VI secretion systems, and these are also associated with activation of the QS system [
119]. In the case of
C. malonaticus, bile salts exposure induced an upregulation of the AcrAB-TolC system, but the molecular mechanisms involved in biofilm formation remain unknown [
120].
When the commensal microbiota species
B. breve and
B. animalis were grown in taurocholic acid or porcine bile, the bacteria bound more effectively to mucin and formed more biofilm but the molecular mechanism is unknown [
121]. Similarly, bile salts can induce biofilm formation in the commensal bacteria
Bacteroides thetaiotaomicron, and this biofilm formation is dependent on the BT3563 DNAse that degrades extracellular DNA in the biofilm matrix [
122].
Human secretory IgA (SlgA) appears to facilitate biofilm formation of the normal gut microbiota in vitro and of
E. coli on the surface of cultured epithelial cells [
123]. SlgA is a key factor that allows agglutination of bacteria and prevents their translocation to the gut epithelial cells, a process known as immune exclusion [
124]. It was observed that mucin facilitated biofilm formation by
E. coli by an unknown mechanism [
123]. Similarly, type-2 mucin increased bacterial adhesion and biofilm formation in
Listeria monocytogenes. This is mediated by the cell-surface protein InlL, which binds directly to Muc-2 [
125]. Mucins are also used by
C. jejuni as a signal to modulate the expression of virulence genes such as mucin degrading-enzymes, flagellin A and toxins [
126]. Moreover,
C jejuni is able to use fucose as a carbon source and shows chemotaxis towards fucose.
C. jejuni biofilm formation decreased in the presence of fucose, suggesting that
C. jejuni in a biofilm is able to coordinate fucose use based on its availability [
127]. Mucus production in the colon is stimulated by the presence of hydrogen sulfide (H
2S), which also promotes the establishment of biofilms in the GIT. H
2S not only promoted biofilm formation by human microbiota ex vivo but also reduced the growth of planktonic bacteria [
128].
Many studies have reported that several hormones and vitamins can affect biofilm formation and subsequent colonization. These factors include peptide hormones, steroid hormones such as catecholamine, and vitamin K [
129]. For example, the hormone epinephrine was found to induce QS in EHEC [
130]. In this study, a
luxS deletion strain, which is unable to produce the EHEC autoinducer AI-3, responded to the host signal epinephrine and activated the expression of genes involved in biofilm formation [
130]. Furthermore,
E. coli biofilm formation is induced by insulin and is increased when glucose is present [
131]. Indeed, the presence of insulin increased
E. coli hydrophobicity and adherence to epithelial cells [
132]. The gut commensal and opportunistic pathogen
Enterococcus faecium can sense and respond to norepinephrine, a human hormone abundant in the gut, by inducing physiological changes, survival and colonization of the host tissues, and biofilm formation [
133]. Catecholamines can also increase adhesion and biofilm formation in the enteropathogens
Salmonella enteritidis and
E. faecalis [
134,
135]. The specific molecular mechanisms of bacterial recognition of the hormones and the activation of regulatory pathways leading to increased biofilm formation have yet to be elucidated. Altogether, these studies show that there is cross-signaling between the host and the microbiota to allow maintenance of the gut homeostasis.
4.2. Antibiotics Affecting Biofilm Formation
Exposure to sub-inhibitory concentrations of antibiotics can induce or inhibit biofilm formation in bacteria. In
E. faecalis, sub-inhibitory concentrations of tigecycline decrease biofilm formation [
136], but sub-inhibitory concentrations of gentamicin significantly increased biofilm formation [
137]. Similarly, sub-inhibitory concentrations of antibiotics that target the cell wall induced biofilm formation in
E. faecalis [
138]. The increase in biofilm formation was associated with an increase in cell lysis, extracellular DNA (eDNA) levels and cell density within the biofilm. This study included a mathematical model that predicted the changes in antibiotic-induced biofilms due to external alterations, showing that perturbations that reduce eDNA or decrease the number of living cells decreased biofilm induction, while compounds that increased cell lysis and cell wall inhibitors increased biofilm formation [
138]. Similar results are also observed in gram-negative bacteria. For example, sub-inhibitory concentrations of aminoglycosides induced biofilm formation in
E. coli [
139]. However, sub-inhibitory concentrations of ceftazidime inhibited
E. coli biofilm formation by increasing the extracellular concentration of indole [
140].
Antibiotic resistance and tolerance can be mediated by efflux pumps and recent studies have suggested that efflux pumps may play a role in biofilm formation [
141]. In
E. coli, efflux pump genes such as
isrA were highly expressed in biofilm bacteria compared to planktonic bacteria [
142]. IsrA mediates the transport of the AI-2 signaling molecule involved in QS, suggesting that efflux pumps may play a role in the transport of the AI in
E. coli biofilms, facilitating QS and promoting biofilm maturation [
143]. Other multidrug efflux pumps such as AcrB and MdtABC were also involved in biofilm formation since corresponding mutant strains decreased biofilm formation and antibiotics resistance [
144,
145]. Similarly, the efflux pumps of
S. enterica play an important role in biofilm formation. Indeed, the inactivation of efflux pumps inhibited the expression of the
S. enterica curli, a surface protein filament that is an essential component of the biofilm matrix [
146]. It was suggested that efflux pumps are involved in the activation of the regulator of curli gene expression [
141]. In
E. coli, some drug-induced stresses repressed production of curli and thus repressed biofilm formation [
147].