When the first cases of HIV infection appeared in the 1980s, AIDS was a deadly disease without any therapeutic alternatives. Currently, there is still no cure for most cases mainly due to the multiple tissues that act as a reservoir for this virus besides the high viral mutagenesis that leads to an antiretroviral drug resistance. Throughout the years, multiple drugs with specific mechanisms of action on distinct targets have been approved.
1. Introduction
The human immunodeficiency virus (HIV) is still a very prominent disease worldwide. Acquired Immunodeficiency Syndrome (AIDS) can now be considered a chronic infection since patients are living longer due to the several options of antiretroviral therapy [1]. The number of new cases has decreased, but nevertheless, according to the latest UNAIDS global statistics, 38.0 million people are living with HIV in 2019 and 1.7 million became newly infected with HIV in 2019 [1,2].
The initial step of HIV replication cycle involves SU binding to the host protein CD4, present in T helper lymphocytes, macrophages, and dendritic cells. The interaction between the CD4 binding site (CD4 bs) present in SU subunit with CD4 causes conformational changes in the SU glycoprotein that exposes the coreceptor binding site [3,4]. Binding to coreceptor induces further conformational changes in the TM subunit, disclosing the fusion peptide which will connect to the target cell membrane due to its extremely hydrophobic nature, thereby enabling the fusion of both viral envelope and host cell membrane [4]. Fusion of viral envelope could occur directly with plasma membrane or alternatively with endosome membrane after endocytosis. Following fusion, the viral capsid is then released in the cytosol forming the reverse transcriptase/pre-integration complex (RTC/PIC) that includes the two copies of gRNA, the viral proteins CA, NC, IN, RT, and Vpr as well as cellular proteins, namely cyclophilin A [5]. Inside this structure, RT converts the single-stranded gRNA into a double stranded DNA (dsDNA). The RTC/PIC is transported through the cytoplasm into the host cell nucleus [5,6] via the nuclear pore, where RTC/PIC interacts with proteins of the nuclear pore complex, namely, Nucleoporin 358 and Nucleoporin153 [5,7]. The structural integrity of RTC/PIC seems to be maintained until it enters the nucleus, minutes before the dsDNA integrates into host cell chromosome [5,6]. The integration process is mediated by the integrase protein (IN) starting by removing nucleotides from the 3′ ends of the proviral DNA, and then, proceeding to catalyze a nucleophilic attack to the phosphodiester bonds of the DNA chains, thus forming a covalent bond between viral and host DNA. This is an essential step in viral replication allowing the establishment of latently infected cells [8,9,10]. Some cell transcription factors enhancers bind to LTR and the regulatory proteins such as Tat, Rev, and Nef are produced. HIV-1 transcription from the LTR promoter is activated by the Tat protein through interaction with the nascent trans -acting-responsive RNA hairpin structure [9,10]. The Env glycoproteins, after translation, processing, and cleavage by cellular furin protease migrate to the plasma membrane. Meanwhile the MA domain targets the Gag-Pol polypeptide to budding sites at the host cell membrane where it interacts with cytoplasmic tail of TM subunit of Env. This event allows virion assembly with gRNA and all the other structural and accessory viral proteins and budding of immature viral particles [11]. During or soon after budding, the viral protease (PR) cleaves the Gag-Pol polypeptide which will allow the release of all structural proteins, such as MA, CA, and NC, as well as their reorganization into mature virions [9,10].
The period between the infection of the first host cell and the detection of the virus in the blood is called the eclipse phase and usually lasts from 7 to 21 days. After the infection of the first cell, the virus continues to replicate in the mucosa, submucosa, and adjacent lymphatic tissue. The replication concentrates in the gut-associated lymphatic tissue (GALT) quite early [12,13]. Then, it follows an exponential rise of the viral loading in which the CD4+ cell counting rapidly decreases. This phase is characterized by flu-like and non-specific clinical signs that usually last between 7 to 10 days. After a few weeks, the immune system can generate a response [9,14]. The cellular immune response starts with the activation of CD8+ cytotoxic lymphocytes. Their T-cell receptor (TCR) will bind to viral proteins, which are in turn connected to the antigen-presenting molecule (MHC I) to eliminate the infected cells [9]. Generally, after 3 to 5 weeks, a humoral response starts to produce specific neutralizing antibodies that will destroy the virions via phagocytosis. The convergence of both types of immune responses leads to a decrease in viremia and a new rise in CD4+ cell count. The period, in which there is an infection but without antibodies, is called the “serological window period” [14]. Even though this is an asymptomatic phase, and the viral loading is somewhat controlled, there is still a loss of immune cells since the virus continues to replicate in the lymphatic tissue (its reservoir), destroying its structure. Then, the viral loading becomes higher as the CD4+ cell count diminishes, leading to the beginning of the AIDS stage. In this stage, the patients are more susceptible to opportunistic infections [14].
Several aspects make it difficult to eradicate the virus once a patient is infected. One of them is related to the absence of proofreading activity in the viral RT, causing a great number of mutations and genetic diversity in the HIV genome. The other one is concerned with the ability of the virus to infect resting memory or naïve cells, leading to a latent viral state [9,14]. The problem with viral latency is that it can occur even after patients have undergone antiretroviral therapy reducing viremia to an undetectable level [9].
2. Novel Therapeutic Strategies
2.1. New Transdermal Drug Delivery Systems
Since most ARVs are administered orally, it is important to identify the main adversities of this route of administration, including those related to bioavailability, frequency of administration, and the hepatic first-pass metabolism. Transdermal drug delivery systems (TDDS) are currently being investigated to diminish those issues [
86,
87,
88,
89]. The TDDS present an alternative to the conventional oral ARV regimens since this administration system allows obtaining a controlled and continuous drug release by avoiding the pharmacokinetic variations of the oral administration. This means that it may be possible to use drugs with a short half-life and find a simpler and acceptable regimen for patients [
86,
87,
89].
Besides the reservoir and the drug itself (i.e., drug concentration, pka, molecular weight (ideally around 500 Da), log P (preferably ranged between 1 and 3), and the melting point) [
86,
87,
88], a TDDS should have other essential components, as follows: (a) permeation enhancers to increase the permeability through the stratum corneum (e.g., pyrrolidones, surfactants, phospholipids, and solvents); (b) pressure-sensitive adhesives (PSA) to keep contact between the skin and the device; (c) a release liner to cover the patch, and (d) a backing layer which should not allow the diffusion of any excipients [
86,
87].
2.1.1. Transdermal Delivery System for Tenofovir Alafenamide
Tenofovir Alafenamide (TAF) is an NRTI commonly used for HIV treatment as part of oral therapy regimens. A controlled matrix delivery system was recently developed for this drug due to its low oral bioavailability [
88]. In this system, the active substance is dispersed in a polymer matrix and a suitable solvent, which later evaporates, forming a drug reservoir. Then, the reservoir is shaped and interconnected with several layers. The overall system is composed of an adhesive layer that controls the release rate of the drug and it is in contact with the skin, followed by the drug reservoir and a second adhesive layer, connected to an exterior impermeable laminate [
86,
87,
88]. Accordingly, this study [
88] aimed to formulate a patch able to continuously release 8 mg/day TAF for one week. For that proposal, silicone-based or polyisobutylene (PIB) suspension patches and acrylate solution patches were used, and several formulations were prepared varying the components, TAF concentration, and permeation enhancers for each patch. Specific parameters were determined, such as (a) crystallization of TAF in each matrix, (b) effect of TAF particle homogenization, (c) coat weight and TAF amount in each patch after exposure to stress conditions, and (d) in vitro skin permeation studies using Franz diffusion cells. Overall, the suspension silicone patch formulated with 15% TAF (
w/
w), silicone, oleic acid, oleyl alcohol, and mineral oil as permeation enhancers revealed to be the most suitable since it presented the target flux permeation rate of 7 µg/cm
2/h through a 50 cm
2 patch area for an entire week, reaching a daily dose of 8.4 mg TAF. Additionally, this formulation was stable over time, non-irritating to the skin, and convenient when peeled off [
88]. Despite these positive results and the promising chance of a new route of administration for TAF, further studies are still needed regarding both pharmacokinetic and safety of this new device [
88].
2.1.2. Transdermal Delivery of Enfuvirtide (T20) via Ultrasounds
As previously discussed, enfuvirtide (T20) is an entry inhibitor, usually administered by subcutaneous injection, 90 mg twice a day, which enables it rather inconvenient in terms of patient compliance. Thus, its transdermal delivery could be quite interesting option. However, there are some obstacles, including the high molecular weight (4.492 Da) of this drug, which could compromise its diffusion through the stratum corneum [
87,
90]. Therefore, the ultrasound technique could be a suitable way to surpass this problem, since it reduces the barrier function of the stratum corneum [
91,
92]. Ultrasound waves are known to create pores that allow large molecules to cross the epidermis besides contributing to a fluid state of lipid skin layers, which in turn, promotes the transcellular pathway [
91,
93].
A recent study has assessed the effects of transdermal delivery of T20 using a low-frequency and low power ultrasound transducer patch in porcine models. The models were divided into 3 separate groups for 30 days as follows: one control group receiving injectable T20 twice a day, another group undergoing ultrasound treatment with saline solution, and a third one treated with transdermal T20 via ultrasounds [
90]. In this last case, T20 was in direct contact with the skin. The final device was obtained using wound dressing patches and a silicone ring that served as a reservoir, over which the ultrasound transducer was placed [
90]. All groups were evaluated to understand the differences between them regarding skin health and bioavailability. The skin health criteria were based on histologic cuts and trans-epidermal water loss. The bioavailability of T20 was evaluated through liquid chromatography/electrospray ionization mass spectrometry (LC-MS/MS) [
90]. Overall, no significant differences between the saline group and the transdermal T20 group were observed, which indicates that ultrasound did not affect the skin. The histologic cuts showed mild signs of inflammation in the active patch group. In addition, the animals from the active patch group had a longer Tmax and a lower Cmax when compared to the injectable T20 group. The plasma concentrations were generally lower with the transdermal treatment as expected [
90].
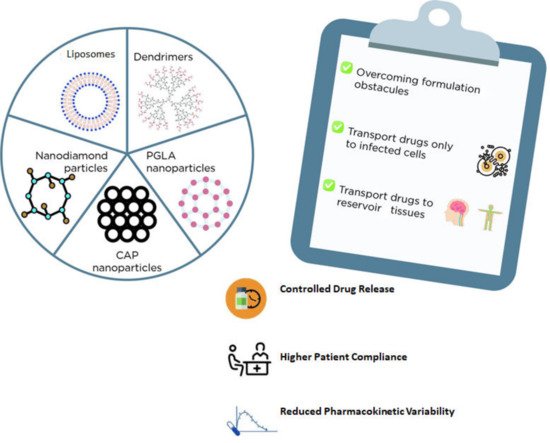
2.2. Nanosystems for Drug Delivery
Nanotechnology has contributed to several applications in drug delivery through different routes of administration and overcoming certain formulation obstacles, such as solubility, bioavailability, and drug stability [
94]. Additionally, nanosystems could be a promising strategy for targeted therapy since they allow the encapsulation of drugs or specific genes that could be transported not only to infected cells but also to reservoir tissues, including the central nervous system and lymph nodes, thereby potentially eradicating the virus [
94].
There are several types of nanosystems that could function as carriers for ARV drugs, such as liposomes, niosomes, solid-lipid or polymeric (e.g., PGLA) or diamond nanoparticles, and dendrimers, among others (
Figure 1) [
94]. In general, liposomes and niosomes are less toxic and cost-effective. Both are made of an aqueous compartment surrounded by a lipid bilayer and could be a promising alternative for drug delivery as these carriers can be easily absorbed by macrophages [
95]. For example, stavudine was already encapsulated into gelatin nanoparticles, which in turn were incorporated into liposomes. Accordingly, several formulations were successfully prepared in this report and characterized in terms of drug release, cytotoxicity, and hemocompatibility [
95].
Figure 1. Examples nanosystems for drug delivery and the main advantages of their usage.
Dendrimers are somewhat toxic but have numerous reactive groups capable of forming conjugates for targeted drug delivery [
94,
96]. Dendrimers possess a typical three-dimensional branched structure where the outer layers are appropriate for conjugation, while the inner layers are quite effective for drug encapsulation, resulting in a controlled drug release [
96]. In a recent study published in 2013, zidovudine was encapsulated into poly (propyl ether imine) dendrimers to surpass the short half-life and provide a more continuous release of this drug [
97]. The study showed that not only this delivery system reduced the hemolytic effect, but it also prolonged the drug release decreasing the occurrence of side effects [
97]. In another study, carbosilane dendrimers (G3-S16 and G2-NF16) were used to encapsulate zidovudine, efavirenz, and tenofovir [
98]. The encapsulated drugs were tested for antiviral activity in PBMC cells and TMZ-bl cells infected with X4 and R5 viral strains. The results showed an enlarged antiviral activity of all three drugs when formulated with dendrimers [
98].
Polymeric nanoparticles, such as quitosan and poly (lactide co-glycolide), are known to be very effective in drug delivery while exhibiting low toxicity like metal gold or silver nanoparticles [
94]. Nanoparticles have been tested to improve ARV delivery to the central nervous system since many ARV drugs suffer efflux mechanisms, which may contribute to the spreading of this virus, and thereby, to the development of HIV-related neurologic disorders [
99,
100]. For example, transferrin is one of the nanoparticles studied for this purpose due to the abundance of transferrin receptors in the blood–brain barrier [
94,
99]. In a recent study, nanodiamond particles were studied to load efavirenz (EFV) and deliver it to the brain [
101]. This study compared nanodiamond particles (ND) with both unmodified and modified surface (ND-COOH and ND-NH2) in terms of toxicity and drug loading capacity. ND-COOH was found to be less suitable than the other two since it induced a higher production of reactive oxygen species (ROS) [
101]. On the contrary, the formulation with unmodified nanodiamond particles (ND-EFV) presented a suitable and slower release profile through a blood–brain barrier model, and it was able to impair viral replication for a longer period in comparison with free EFV [
101]. Overall, this study suggests that ND particles are a promising drug delivery system, due to their nontoxic nature and ability to cross the blood–brain barrier. However, further studies still need to be performed to evaluate the effect in in vivo models and possible side effects, since ND particles may interfere with the expression of genes related to neuronal function [
101]. Another recent study attempted to demonstrate the effects of using PGLA nanoparticles loaded with EFV and saquinavir (SQV) [
102]. In general, ARV formulated with PGLA nanoparticles showed lower IC50 values in comparison with the free ARV drugs. The release profile of these nanoparticles was also quite favorable, since the drugs were first rapidly released, and then, at a continuous rate. Additionally, when adding free tenofovir to these nanoparticles, a synergic effect was obtained resulting in dose reduction to impair HIV activity [
99]. In another report, PGLA nanoparticles were loaded with raltegravir (RAL) and EFV, and further incorporated in a thermo-sensitive vaginal gel for HIV prophylaxis [
103]. The goal was to prepare a gel that would acquire a gel-like texture at 37 °C and liquid at room temperature. The PGLA nanoparticles were here prepared via a modified emulsion-solvent evaporation method and characterized through multiple experiments, including in vitro release studies with human cervical cells. When compared with an EFV-RAL solution, the loaded nanoparticles had a lower EC90 and were able to ensure a constant release of these drugs, despite their different intracellular concentrations and routes of metabolization [
103]. Overall, this formulation was considered a successful and promising option for pre-exposure HIV prophylaxis [
103]. Still regarding prophylaxis, cellulose acetate-phthalate (CAP) nanoparticles have shown promising outcomes when incorporated into thermosensitive gels. Although most used as a coating agent for other formulations, CAP was found to possess ARV activity by promoting viral disintegration and interfering with the mechanisms of viral entry. Moreover, CAP was stable at low pH, which facilitated a vaginal drug delivery [
104,
105]. In another study [
105], a thermosensitive gel was formulated using EFV-loaded CAP nanoparticles. This formulation was assessed for cytotoxicity and prophylactic activity in human cervical cells (HeLa) and TZM-bl cells against an EFV solution. The results revealed a remarkable encapsulation efficacy as well as lower cytotoxicity in HeLa cells treated with EFV-CAP nanoparticles formulated in the thermosensitive gel. Moreover, these nanoparticles showed enhanced prophylactic activity (at 5 ng/mL) in TMZ-bl cells when compared with the EFV solution [
105].
Similarly, dolutegravir (DTG) loaded CAP nanoparticles were incorporated into a thermosensitive gel and tested at pH = 4.2 and pH = 7.4 to simulate vaginal and seminal fluid conditions, respectively [
104]. In this study, the pH clearly influenced both the drug release and the cytotoxicity of the tested formulation [
104].
A novel tenofovir alafenamide fumarate (TAF) nanofluidic implant developed by Pons-Faudoa et al. [
105,
106] revealed to be potential as a subcutaneous delivery platform for long-term pre-exposure prophylaxis to HIV transmission according to in vivo data (SHIV-Challenged Nonhuman Primates). It had similar effects as oral TAF dosing with a lower dose [
105].
In summary, the examples of the most recent in vitro studies of ARV loaded nanocarriers for HIV management are listed in
Table 3. Overall, the use of long-acting ARVs with alternative drug delivery systems contributes for the long-term sustained release of these drugs with a controlled kinetic profile which certainly accounts for the success of these new ARVs therapies. In addition, the drug delivery systems might also provide more convenient dosing, enhance tissue penetrance, improve viral resistance profile, and reduce drug toxicity [
105].
Table 3. Examples of main outcomes from in vitro studies of ARV loaded nanocarriers for HIV management.
This entry is adapted from the peer-reviewed paper 10.3390/molecules26175305