Bile acids (BA) are amphipathic steroid acids synthesized from cholesterol in the liver. They act as detergents to expedite the digestion and absorption of dietary lipids and lipophilic vitamins. BA are also considered to be signaling molecules, being ligands of nuclear and cell-surface receptors, including farnesoid X receptor and Takeda G-protein receptor 5. Moreover, BA also activate ion channels, including the bile acid-sensitive ion channel and epithelial Na+ channel. BA regulate glucose and lipid metabolism by activating these receptors in peripheral tissues, such as the liver and brown and white adipose tissue. Recently, 20 different BA have been identified in the central nervous system. Furthermore, BA affect the function of neurotransmitter receptors, such as the muscarinic acetylcholine receptor and γ-aminobutyric acid receptor. BA are also known to be protective against neurodegeneration.
- bile acids
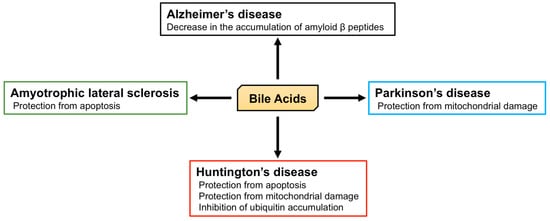
- Alzheimer’s disease
- Parkinson’s disease
- Huntington’s disease
- ALS
1. Introduction
2. Neurological Functions of BA
2.1. The Role of BA in the Brain
2.2. The Role of BA in Neurodegenerative Diseases
This entry is adapted from the peer-reviewed paper 10.3390/biom9060232
References
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693.
- Hofmann, A.F.; Hagey, L.R.; Krasowski, M.D. Bile salts of vertebrates: Structural variation and possible evolutionary significance. J. Lipid Res. 2010, 51, 226–246.
- Reschly, E.J.; Ai, N.; Ekins, S.; Welsh, W.J.; Hagey, L.R.; Hofmann, A.F.; Krasowski, M.D. Evolution of the bile salt nuclear receptor FXR in vertebrates. J. Lipid Res. 2008, 49, 1577–1587.
- Abrams, P.; Andersson, K.E.; Buccafusco, J.J.; Chapple, C.; de Groat, W.C.; Fryer, A.D.; Kay, G.; Laties, A.; Nathanson, N.M.; Pasricha, P.J.; et al. Muscarinic receptors: Their distribution and function in body systems, and the implications for treating overactive bladder. Br. J. Pharmacol. 2006, 148, 565–578.
- Kiriyama, Y.; Nochi, H. D-Amino Acids in the Nervous and Endocrine Systems. Scientifica (Cairo) 2016, 2016, 6494621.
- Montes de Oca Balderas, P. Flux-Independent NMDAR Signaling: Molecular Mediators, Cellular Functions, and Complexities. Int. J. Mol. Sci. 2018, 19, 3800.
- Kumar, A. NMDA Receptor Function During Senescence: Implication on Cognitive Performance. Front. Neurosci. 2015, 9, 473.
- Cicek, S.S. Structure-Dependent Activity of Natural GABA(A) Receptor Modulators. Molecules 2018, 23, 1512.
- Schubring, S.R.; Fleischer, W.; Lin, J.S.; Haas, H.L.; Sergeeva, O.A. The bile steroid chenodeoxycholate is a potent antagonist at NMDA and GABA(A) receptors. Neurosci. Lett. 2012, 506, 322–326.
- Fujita, A.; Bonnavion, P.; Wilson, M.H.; Mickelsen, L.E.; Bloit, J.; de Lecea, L.; Jackson, A.C. Hypothalamic Tuberomammillary Nucleus Neurons: Electrophysiological Diversity and Essential Role in Arousal Stability. J. Neurosci. 2017, 37, 9574–9592.
- Xie, J.F.; Fan, K.; Wang, C.; Xie, P.; Hou, M.; Xin, L.; Cui, G.F.; Wang, L.X.; Shao, Y.F.; Hou, Y.P. Inactivation of the Tuberomammillary Nucleus by GABAA Receptor Agonist Promotes Slow Wave Sleep in Freely Moving Rats and Histamine-Treated Rats. Neurochem. Res. 2017, 42, 2314–2325.
- Yanovsky, Y.; Schubring, S.R.; Yao, Q.; Zhao, Y.; Li, S.; May, A.; Haas, H.L.; Lin, J.S.; Sergeeva, O.A. Waking action of ursodeoxycholic acid (UDCA) involves histamine and GABAA receptor block. PLoS ONE 2012, 7, e42512.
- Soares, R.; Ribeiro, F.F.; Xapelli, S.; Genebra, T.; Ribeiro, M.F.; Sebastiao, A.M.; Rodrigues, C.M.P.; Sola, S. Tauroursodeoxycholic Acid Enhances Mitochondrial Biogenesis, Neural Stem Cell Pool, and Early Neurogenesis in Adult Rats. Mol. Neurobiol. 2018, 55, 3725–3738.
- Bond, A.M.; Ming, G.L.; Song, H. Adult Mammalian Neural Stem Cells and Neurogenesis: Five Decades Later. Cell Stem Cell 2015, 17, 385–395.
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71.
- Cole, S.L.; Vassar, R. The Basic Biology of BACE1: A Key Therapeutic Target for Alzheimer’s Disease. Curr. Genom. 2007, 8, 509–530.
- Tomita, T. Molecular mechanism of intramembrane proteolysis by gamma-secretase. J. Biochem. 2014, 156, 195–201.
- Sun, L.; Zhao, L.; Yang, G.; Yan, C.; Zhou, R.; Zhou, X.; Xie, T.; Zhao, Y.; Wu, S.; Li, X.; et al. Structural basis of human gamma-secretase assembly. Proc. Natl. Acad. Sci. USA 2015, 112, 6003–6008.
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158.
- Nunes, A.F.; Amaral, J.D.; Lo, A.C.; Fonseca, M.B.; Viana, R.J.; Callaerts-Vegh, Z.; D’Hooge, R.; Rodrigues, C.M. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-beta deposition in APP/PS1 mice. Mol. Neurobiol. 2012, 45, 440–454.
- Lo, A.C.; Callaerts-Vegh, Z.; Nunes, A.F.; Rodrigues, C.M.; D’Hooge, R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 2013, 50, 21–29.
- Pan, X.; Elliott, C.T.; McGuinness, B.; Passmore, P.; Kehoe, P.G.; Holscher, C.; McClean, P.L.; Graham, S.F.; Green, B.D. Metabolomic Profiling of Bile Acids in Clinical and Experimental Samples of Alzheimer’s Disease. Metabolites 2017, 7, 28.
- Marksteiner, J.; Blasko, I.; Kemmler, G.; Koal, T.; Humpel, C. Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in Alzheimer’s disease. Metabolomics 2018, 14, 1.
- MahmoudianDehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease-An emerging role for gut microbiome. Alzheimers Dement. 2019, 15, 76–92.
- Area-Gomez, E.; de Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018, 9, 335.
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants (Basel) 2017, 6, 25.
- Kiriyama, Y.; Nochi, H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2018, 7, 1.
- Bell, S.M.; Barnes, K.; Clemmens, H.; Al-Rafiah, A.R.; Al-Ofi, E.A.; Leech, V.; Bandmann, O.; Shaw, P.J.; Blackburn, D.J.; Ferraiuolo, L.; et al. Ursodeoxycholic Acid Improves Mitochondrial Function and Redistributes Drp1 in Fibroblasts from Patients with Either Sporadic or Familial Alzheimer’s Disease. J. Mol. Biol 2018, 430, 3942–3953.
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376.
- Kiriyama, Y.; Nochi, H. The Function of Autophagy in Neurodegenerative Diseases. Int. J. Mol. Sci. 2015, 16, 26797–26812.
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414.
- Hardy, J. Genetic analysis of pathways to Parkinson disease. Neuron 2010, 68, 201–206.
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273.
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol. Cell 2015, 60, 7–20.
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314.
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934.
- Connolly, N.M.C.; Theurey, P.; Adam-Vizi, V.; Bazan, N.G.; Bernardi, P.; Bolanos, J.P.; Culmsee, C.; Dawson, V.L.; Deshmukh, M.; Duchen, M.R.; et al. Guidelines on experimental methods to assess mitochondrial dysfunction in cellular models of neurodegenerative diseases. Cell Death Differ. 2018, 25, 542–572.
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.J.; Wolf, C.R.; Rodrigues, C.M.; Gama, M.J. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson’s disease. Mol. Neurobiol. 2012, 46, 475–486.
- Moreira, S.; Fonseca, I.; Nunes, M.J.; Rosa, A.; Lemos, L.; Rodrigues, E.; Carvalho, A.N.; Outeiro, T.F.; Rodrigues, C.M.P.; Gama, M.J.; et al. Nrf2 activation by tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Exp. Neurol. 2017, 295, 77–87.
- Rosa, A.I.; Duarte-Silva, S.; Silva-Fernandes, A.; Nunes, M.J.; Carvalho, A.N.; Rodrigues, E.; Gama, M.J.; Rodrigues, C.M.P.; Maciel, P.; Castro-Caldas, M. Tauroursodeoxycholic Acid Improves Motor Symptoms in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 9139–9155.
- Rosa, A.I.; Fonseca, I.; Nunes, M.J.; Moreira, S.; Rodrigues, E.; Carvalho, A.N.; Rodrigues, C.M.P.; Gama, M.J.; Castro-Caldas, M. Novel insights into the antioxidant role of tauroursodeoxycholic acid in experimental models of Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2171–2181.
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Mol. Neurobiol. 2016, 53, 810–817.
- Dayalu, P.; Albin, R.L. Huntington disease: Pathogenesis and treatment. Neurol. Clin. 2015, 33, 101–114.
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983.
- Tunez, I.; Tasset, I.; Perez-De La Cruz, V.; Santamaria, A. 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: Past, present and future. Molecules 2010, 15, 878–916.
- Li, J.Y.; Popovic, N.; Brundin, P. The use of the R6 transgenic mouse models of Huntington’s disease in attempts to develop novel therapeutic strategies. NeuroRx 2005, 2, 447–464.
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Linehan-Stieers, C.; Abt, A.; Kren, B.T.; Steer, C.J.; Low, W.C. A bile acid protects against motor and cognitive deficits and reduces striatal degeneration in the 3-nitropropionic acid model of Huntington’s disease. Exp. Neurol. 2001, 171, 351–360.
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506.
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676.
- Juenemann, K.; Jansen, A.H.P.; van Riel, L.; Merkx, R.; Mulder, M.P.C.; An, H.; Statsyuk, A.; Kirstein, J.; Ovaa, H.; Reits, E.A. Dynamic recruitment of ubiquitin to mutant huntingtin inclusion bodies. Sci. Rep. 2018, 8, 1405.
- Mancuso, R.; Navarro, X. Amyotrophic lateral sclerosis: Current perspectives from basic research to the clinic. Prog. Neurobiol. 2015, 133, 1–26.
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23.
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech 2019, 12.
- Muyderman, H.; Hutson, P.G.; Matusica, D.; Rogers, M.L.; Rush, R.A. The human G93A-superoxide dismutase-1 mutation, mitochondrial glutathione and apoptotic cell death. Neurochem. Res. 2009, 34, 1847–1856.
- Vaz, A.R.; Cunha, C.; Gomes, C.; Schmucki, N.; Barbosa, M.; Brites, D. Glycoursodeoxycholic acid reduces matrix metalloproteinase-9 and caspase-9 activation in a cellular model of superoxide dismutase-1 neurodegeneration. Mol. Neurobiol. 2015, 51, 864–877.
- Elia, A.E.; Lalli, S.; Monsurro, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2016, 23, 45–52.