The NLRP3 inflammasome (NOD-, LRR-, and pyrin domain-containing protein 3) is an intracellular, multiprotein signalling complex implicated in a plethora of inflammatory diseases. These integral elements include the sensor NLRP3 protein, an adaptor protein called adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC) and procaspase-1. The sensor NLRP3 protein can be triggered by PAMPs, DAMPs, and a range of diverse external stimuli such as infection and injury.
- NLRP3 inflammasome
- glaucoma
- RGC (retinal ganglion cells)
- inflammation
1. Introduction
Glaucoma is a neurodegenerative disease and the leading cause of irreversible blindness worldwide. Glaucoma affects more than 70 million people, 10% of which are bilaterally blind [1,2]. The prevalence of glaucoma is estimated to increase to 111.8 million by 2040, which can be attributed to an aging population [3]. Glaucoma is characterised by degeneration of the retinal ganglion cell (RGC) axons resulting in damage or remodelling of the optic nerve head (ONH), as evidenced by the characteristic clinical sign of optic disc cupping [4]. These clinical manifestations lead to disruption of the visual pathway and vision loss in one or both eyes [5]. The most common type of glaucoma is primary open-angle glaucoma (POAG)—referred to as “glaucoma” in this entry. POAG is characterised by diminished outflow of aqueous humour (AH) despite an unobstructed or open irideocorneal angle [6]. Many patients with glaucoma present with elevated intraocular pressure (IOP > 21 mmHg); however, this is not a requirement for the diagnosis of glaucoma, with 30–40% presenting with normal IOP [5].
The pathophysiology of glaucoma is not well understood and, currently, IOP is the only modifiable risk factor in the disease [7]. The goal of interventions for glaucoma are to lower IOP to a level that may prevent further damage to the ONH and therefore vision loss [5,6,7,8]. Interventions include topical medications that can be applied to the ocular surface, oral medications, laser treatment, and surgery to regulate AH outflow and production to lower IOP [1]. However, approximately 30–40% of POAG patients exhibit a normal IOP, indicating that elevated IOP is not the sole causative factor of glaucoma [9]. In fact a number of risk factors, such as genetics, age, and lifestyle, trigger common pathological endpoints resulting in glaucomatous optic neuropathy [10].
Impaired axonal transport in the RGCs, observed in both animal and human studies, is reported as a potential mechanism of damage in glaucoma [11]. Ischemia, restriction of blood to the bodies tissues, has also been implicated in the pathogenesis of glaucoma, with reduced ONH blood flow reported in glaucoma patients [12]. Excitotoxicity, which results in neuron damage, has been an area of interest in the pathophysiology of glaucoma; however, the data in this area are contradictory [5,13]. Moreover, there is an emerging body of evidence to support the role of inflammation in glaucoma pathogenesis [14,15].
2. The NLRP3 Inflammasome
Pattern recognition receptors (PRRs) recognise danger signals, such as, pathogen associated molecular patterns (PAMPs) and danger associated molecular patterns (DAMPs) [53]. Nod like receptors (NLRs) are a type of PRR with a nucleotide-binding and oligomerisation domain, and act as receptors in the cytoplasm [50]. NLRs are categorised into four subgroups based on their N-terminal domain, with the NLRP group containing a pyrin domain [54]. NLRP3 is one member of the NLRP family, of which there are 14 members, all of which are involved in the formation of inflammasomes [55]. NLRP3 is the most well characterised member of the inflammasome family [56], and is an important regulator of inflammatory diseases and plays a key role in the innate immune system [57].
The NLRP3 inflammasome (NOD-, LRR-, and pyrin domain-containing protein 3) is an intracellular, multiprotein signalling complex implicated in a plethora of inflammatory diseases [55]. These integral elements include the sensor NLRP3 protein, an adaptor protein called adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC) and procaspase-1 [58]. The sensor NLRP3 protein can be triggered by PAMPs, DAMPs, and a range of diverse external stimuli such as infection and injury. NLRP3 inflammasome activation is a tightly regulated system and requires two signals for activation, as depicted in Figure 1. The priming signal results in an increased expression of NLRP3, pro-IL-1β, and pro-IL-18, as the nuclear factor-kappa beta (NF-κβ) pathway is activated in response to a stimulus [58]. The priming step of inflammasome activation is tightly controlled by a series of different post translational modifications of NLRP3, ASC, and caspase-1, including ubiquitination, phosphorylation, and sumoylation [59].
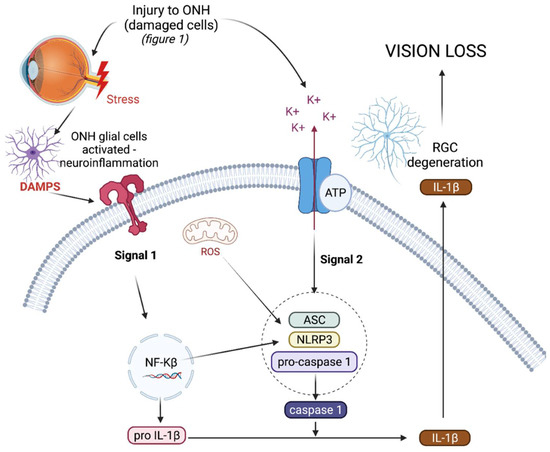
Triggering or activation is the second signal needed for inflammasome activation, and involves the oligomerisation of NLRP3 in its inactive form with procaspase-1 and ASC. Generally the activation step of inflammasome oligomerisation is a result of potassium efflux from the cell [59]. This results in the cleavage of procaspase-1 into caspase-1 and, as a result, pro-IL-1β and pro-IL-18 into active IL-1ß and IL-18, which are subsequently secreted from the cell [60,61]. As well as producing pro-inflammatory cytokines, active caspase-1 also results in the cleavage of gasdermin-D (GSDMD), a pore forming protein, resulting in pyroptosis, a form of cell death [62]. In the chronic diseases driven by inflammation, such as type 2 diabetes and cryopyrin-associated periodic syndromes (CAPS), the NLRP3 inflammasome is dysregulated, resulting in the uncontrolled release of pro-inflammatory cytokine IL-1β, which drives inflammation in such diseases [63,64].
3. The NLRP3 Inflammasome in Glaucoma
Dysregulation of the NLRP3 inflammasome has been implicated in several neurodegenerative diseases, including Alzheimer’s disease and multiple sclerosis [85]. Increased levels of IL-1β mRNA and protein have also been observed in the blood of glaucoma patients compared with controls, suggesting activation of the NLRP3 inflammasome in glaucoma [35]. Furthermore, activation of the NLRP3 inflammasome has been associated with the induction of IL-1β and death of RGCs in mouse models of acute glaucoma via optic nerve (ON) crush. [64,86]. In an inducible mouse model of glaucoma, the use of fluorescent reporter mice to track inflammasome activation demonstrated that NLRP3 inflammasome activation occurs early in the ONH, following IOP elevation, and coincides with the induction of pro-inflammatory cytokines and Iba1+, a microglia marker, immune cells in the ONH [87]. This upregulation of inflammatory genes occurs as early as 7 days post IOP elevation in the ONH and before the induction of inflammatory genes occurs in the retina [79]. With the use of knockout mice lacking various components of the NLRP3 inflammasome, we can conclude that the NLRP3 inflammasome activation is required for RGC death [64,79,88]. These studies support the hypothesis that “danger” signals in the eye, IOP elevation being one, activates the NLRP3 inflammasome pathway in the glial cells of the ONH and retina, resulting in neurotoxic inflammation, axon degeneration, and subsequent death of RGCs in glaucoma models.
4. NLRP3 Inflammasome as a Target for Therapy in Glaucoma
There is an obvious need for the development of new medications for glaucoma, given that current treatments work to lower IOP only. However, these treatments are not successful in all patients [110,111]. NLRP3 may be a promising IOP independent target for the treatment of glaucoma [112]. Several studies demonstrate that the inhibition of NLRP3 activation significantly inhibits the death of RGCs in experimental models of retinal ischemia/reperfusion injury (acute glaucoma) [113,114,115,116]. Blockade of the P2x7r, which can act as signal 2 in inflammasome activation with A438079, a P2x7r inhibitor, also attenuates RGC death by inhibiting inflammasome activation. Additionally in an in-vitro model, ATP, which binds to the P2x7r, was shown to induce inflammation in the retina by activating the NLRP3 inflammasome [76]. High-mobility group box 1 (HMGB1), which is often released from damaged cells, is known to be released upon rapid IOP elevation and is also a known DAMP that activates Toll Like Receptor 4 (TLR-4) to activate the NF-kB pathway. In an acute glaucoma mouse model, the release of HMGB1 resulted in increased levels of NLRP3 and IL-1B. Inhibition of HMGB1 resulted in reduced NLRP3 and IL-1B levels, which also reduced RGC death and glaucoma severity [117].
However, in the more clinically relevant microbead-induced mouse model of glaucoma, where IOP was elevated, treatment with a commercially available NLRP3-specific inhibitor (MCC950) [118] was also shown to prevent axon degeneration and death of RGCs [119]. In this microbead-induced model of glaucoma, 4 weeks of elevated IOP resulted in a 25–30% loss of RGCs, which was significantly attenuated following a three-times a week treatment with intraperitoneal MCC950. Mice treated with MCC950 displayed axon and RGC densities equal to that of the non-glaucoma controls. MCC950 injection, however, had no effect on IOP elevation in the treated mice [119]. Currently, this is the only study that has evaluated a NLRP3-specific inhibitor in an inducible model of glaucoma [119]. Collectively, these studies provide evidence that NLRP3 inhibition may be a novel therapeutic strategy to protect RGCs and prevent axon degeneration in glaucoma.
MCC950, a small molecule drug, however, does present with some drawbacks, including the very short half-life of MCC950 of only 3 h [120]. This means the drug must be administered regularly either systemically or by multi-intravitreal injections, both of which are not desirable for diseases of the eye. Other limitations of small molecule inhibitors include that they are often not fully characterised, and many are not completely specific to their targets [121,122]. Therefore, there has been interest in recent years in the development of biologics, such as antibodies for the treatment of many chronic conditions, as they have revolutionised the treatment and modification of many diseases including some cancers and autoimmune rheumatic diseases [121,122]. Biologics are well known to have a longer half-life, allowing for longer dosing intervals because of their larger size, which is an important factor in the treatment of glaucoma [122,123]; they are also recycled by the body and have a high affinity and potency. The major advantage of biologic molecules for treatment is the specificity of their mechanisms of actions preventing off target effects [122]. Financial cost in the long run is also an advantage of biologic therapies, as they are expected to deliver a better economic return than small molecules [121]. The use and approval of biologics in medicine is increasing each year, targeting extracellular or cell membrane proteins [123]. However, the delivery mechanisms of antibodies into cells to target intracellular molecules is a major obstacle [123]. It is important to note that, generally, biologics targeting the NLRP3/IL-1β pathway target secreted IL-1β directly, like Canakinumab to suppress its inflammatory responses, which poses a serious risk of fatal infection, as IL-1β cannot be produced in response to an infection [49]. Targeting NLRP3 to supress IL-1β production means IL-1β can still be produced by other pathways if the body encounters an acute infection [124]. Anakinra is another biologic that targets the NLRP3 pathway binds to IL-1R1 to inhibit IL-1β binding to IL-1R1, as this can further activate the NLRP3 inflammasome [125]. There is a number of small molecule inhibitors that target NLRP3 to suppress its effects or inhibit NLRP3 inflammasome oligomerisation [114,126,127,128,129].
This entry is adapted from the peer-reviewed paper 10.3390/biom11081239