1. Introduction
γ-
l-glutamyl-
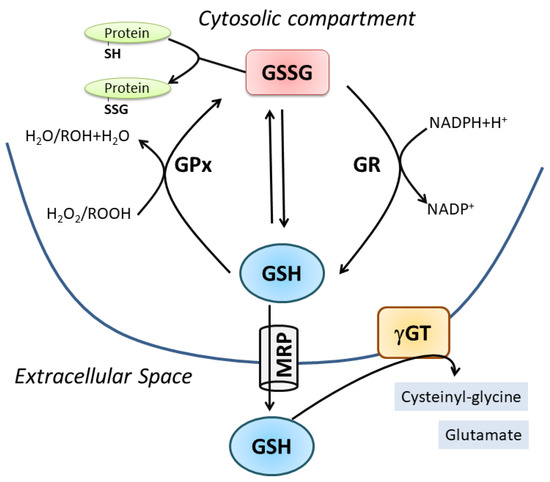
l-cysteinyl-glycine (glutathione, GSH) is the most versatile non-enzymatic antioxidant involved in cell signaling, detoxification and oxy-radical scavenging activity. Its structure, cellular concentration and the systems deputed to its synthesis, degradation and regeneration concur to these different functions. For instance: (i) GSH synthesis is a two-step process mediated by cytosolic enzymes (a faster process with respect to a gene-mediated one); (ii) the peculiar iso-peptide bond of the glutamate makes GSH a protease-resistant peptide; (iii) the NADPH-reductase systems guarantee the maintaining of a high ratio between reduced (GSH) and oxidized (GSSG, GSSR) forms under physiological conditions. Alteration of such ratio during oxidative burst can modulate redox signaling pathways related to various cellular processes including proliferation, growth and apoptosis [
1].
Two ATP-dependent cytosolic enzymes are responsible for the de novo synthesis of GSH: glutamate-cysteine ligase (GCL) also named γ-glutamylcysteine synthase (GCS) and glutathione synthase (GS). The first is the rate-limiting enzyme formed by a modulatory or light subunit (GCLM) and a catalytic or heavy subunit (GCLC). Using ATP hydrolysis, GCLC is able to shape an iso-peptide bond between the γ-carboxyl of glutamate and the amino group of cysteine. GS catalyzes the addition of glycine to γ-glutamylcysteine created by GCLC to form GSH [
1]. GCLC is transcriptionally regulated by Nuclear factor (erythroid-derived 2)-like 2 (NFE2L2), a transcription factor that regulates a wide array of antioxidant responsive element-driven genes in various cell types [
2]. GSH degradation occurs in the extracellular space by the enzyme γ-glutamyl-transpeptidase (γ-GT), which cleaves GSH into cysteinyl-glycine (Cys-Gly) and glutamate. Thus, systemic GSH homeostasis relies on its intracellular synthesis, export and degradation outside the cell in a cycle with a half-life of 2–3 h [
3]. Intracellularly, GSH is present in a dynamic equilibrium with its oxidized forms, mainly GSSG, which is the product of a disulfide bridge between two molecules of GSH directly produced upon oxidative burst or by the activity of the antioxidant enzyme glutathione peroxidase (GPx). Subsequently, GSSG is converter to GSH through the glutathione reductase (GR). GSSR represents a less concentrated form deriving from mixed disulfide formation with protein thiols (protein S-glutathionylation); however, this oxidized form is pivotal in redox signaling pathways representing a reversible post-translational modification of proteins [
4]. The value of GSH/GSSG ratio is universally accepted as an index of cell redox status because of the scavenging effect of GSH against several reactive oxygen species (e.g., hydroxyl radical: OH, superoxide anion: O
2−, hydrogen peroxide: H
2O
2, lipid peroxyl radical: HOO), either directly or through the activity of antioxidant enzymes, mainly the glutathione peroxidases. GSH homeostasis is fundamental for cell metabolism and function as stated by the association of altered GSH levels with numerous conditions and pathologies such as aging, neurodegeneration, cancer, inflammation and muscle degeneration (
Figure 1) [
5,
6,
7,
8].
Figure 1. Cellular GSH homeostasis. For details see the text.
GSH plays an important role also in the field of nitric oxide (NO) physiology and pathology [
9,
10,
11]. NO is a gaseous free radical synthesized by NO synthases (NOSs) starting from L-arginine, NADPH and oxygen. NO is a highly reactive molecule that despite being potentially toxic is implicated in a wide range of physiological processes, such as neurotransmission, vascular tone regulation, musculature contraction/relaxation and platelet aggregation [
12,
13,
14]. Furthermore, it is an integral part of the inflammatory response to pathogens and cancer cells [
15,
16]. This versatility of NO mainly derives from its complex chemistry. Depending on its distribution and reactants, NO can exist in different redox forms with distinct and highly reactive properties, such as nitroxyl (HNO), oxides (NO
2, N
2O
4 and N
2O
3), peroxynitrite (ONOO
·−) and S-nitrosothiols (RSNO) [
17].
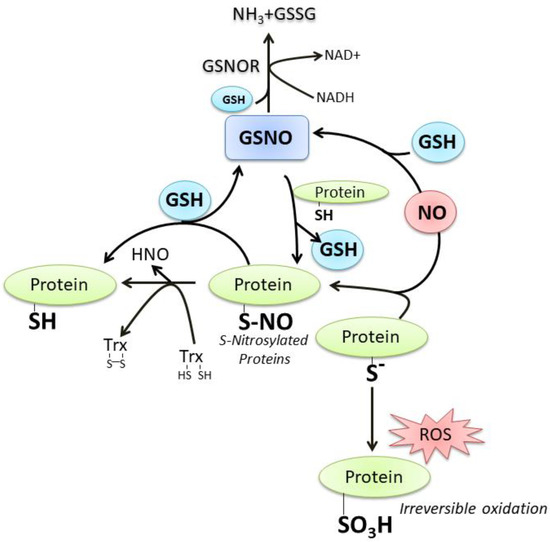
GSH and NO can easily interact at different levels; a number of still debated mechanisms commit NO to react with GSH with the formation of S-nitrosoglutathione (GSNO), which is considered an endogenous NO reserve that contributes to the intracellular regulation of nitrosative stress [
18]. In fact, we and other researchers demonstrated that neuronal NOS (nNOS) overexpression or treatments with NO donors result in intracellular GSH increase, a process fundamental to counteract neuronal death [
11,
19,
20]. Thiol of “reactive cysteine” on proteins (P-SH) is another important target of reactive oxygen/nitrogen species (ROS/RNS) signaling pathway because it can be oxidized at a different extent up to the sulfonic form (P-SO
3H), an irreversible oxidation product. On the contrary, the sulfenic form (P-SOH) represents the most frequent and reversible modification under physiological ROS/RNS flux. It can react with NO to produce the S-nitrosylation derivative (P-SNO), which seems to be implicated in the majority of long-range NO-cellular redox processes. The P-SNO can be reconverted to P-SH by thiol-dependent systems, among which GSH plays a prominent role through the translocation reaction of NO that generates GSNO [
18,
21] (
Figure 2). Based on this evidence, it is conceivable to link the majority of NO effects on the availability and redox status of GSH.
Figure 2. The crosstalk between GSH and NO. For details see the text.
2. Oxidative/Nitrosative-Mediated Signaling and Stress as the Conditions Where NO Mainly Intersects GSH
Oxidative/nitrosative stress is properly “an imbalance between oxidants and antioxidants in favor of the oxidant species, potentially leading to damage” [
26,
27]. ROS are by-products of the sequential one-electron reduction of molecular oxygen, which includes radical and non-radical species. RNS are a group of reactive compounds that include NO, which is physiologically produced by NOSs, a NADPH-dependent family of enzymes present in most cells and tissues [
28]. To date, three main isoenzymes belonging to this family have been cloned and purified: nNOS, endothelial NOS (eNOS) and inducible NOS (iNOS) [
29]. The first two are constitutively expressed and calcium (Ca
2+)-dependent enzymes. nNOS is mainly expressed in cells of neuronal origin where NO functions as a neuromodulator and neurotransmitter in the central and peripheral nervous system. Notably, the nNOS isoform is also present at the skeletal and cardiac muscle level, namely the nNOSμ [
30,
31]. It is characterized by a 34-amino acid insert that arises from the alternative splicing of nNOS pre-mRNA between exons 16 and 17. The catalytic activity is similar to that of nNOS and its expression coincides with the formation of differentiated myotube in cultured cells. eNOS is localized in the endothelium, in cardiomyocytes and adipocytes. It principally participates in the regulation of blood pressure and vascular tone. Instead, iNOS is a Ca
2+-independent and cytokine-inducible enzyme, expressed in macrophages during inflammation and tissue injury [
32]. In recent years, the importance of the fourth isoform of NOS has also emerged: the mitochondrial NOS (mtNOS) [
33]. It is localized in mitochondria of several tissues, including skeletal muscle, where it regulates O
2 consumption [
34].
NO is a hydrophobic gas molecule with a high diffusion coefficient but a short lifetime mainly due to its rapid reaction with oxygen and multiple cellular components. Therefore, as in the case of ROS, low and regulated production of NO is associated with paracrine signaling processes whereas its overproduction can cause damage to biological macromolecules and determine many pathological states [
18,
23,
35]. In this regard, the product of NO reaction with O
2− leads to the formation of ONOO
·−, which is a strong oxidant that reacts with lipids, DNA, and proteins via direct oxidative reactions or via indirect, radical-mediated mechanisms [
36].
3. GSH/NO Crosstalk in Skeletal Muscle Physiology
Skeletal muscle represents the most important district of the body where fluctuations of ROS/RNS could be observed in relation to physiological function. Muscle cell homeostasis is strictly dependent on regulated NO production by nNOS localized at the acetylcholine receptor [
47]. Consistently, NO is involved in myotube differentiation and synaptic signaling from nerve to myotube. An increase in ROS production also occurs during differentiation due to the dramatic increase of NADPH oxidases activity [
48]. On the other side, myoblasts derived from GPx1 knockout (Gpx1-/-) mice scarcely differentiate leading to only a few myotubes with respect to wild-type cells [
49].
Physical activity of the skeletal muscle is directly correlated with O
2 consumption, therefore, more intense is the exercise greater is the production of ROS/RNS. Moreover, such changes could be very fast due to conversion from rest to intense muscle contraction. This could explain the various and efficient antioxidant systems at the muscle level including various isoenzymes of Superoxide dismutase (SOD), GPx and catalase, high activity of GSH-dependent enzymes and a strong ability to synthesize new GSH [
24,
50]. Additionally, GSH levels in skeletal muscle are related to the metabolic profile of the tissue; in healthy human skeletal muscle fibers, the level of GSH is higher in aerobic type I fibers than in anaerobic type II fibers [
51]. There are several potential sources of ROS/RNS in skeletal muscle but also other organs can participate in their production during exercise. However, because of the invasive nature of analyses used to determine antioxidants variations, the majority of the studies during exercise in humans evaluated blood markers, whereas data at muscle level were mainly from animals exposed to exercise training. As generally accepted, mitochondria represent the major source of ROS, but this is still debated
during exercise at skeletal muscle since mitochondria produce more ROS under state 4 (basal) with respect to state 3 (maximum rate of ADP-stimulated respiration), the latter being the condition under aerobic muscle contraction [52]. Additional sources, as above mentioned, include NADPH-oxidase enzymes located at the different membrane structures (sarcoplasmic reticulum, plasma membrane, transverse tubule), phospholipase A2 and xanthine oxidase [50]. Changes in expression/activity of antioxidant enzymes, such as SOD and GPx, were deeply investigated both in humans and animals during training; their increases are muscle-specific with higher effect in oxidative fibers also at mitochondrial level [24,51]. Controversial results were reported for catalase activity because the majority of the studies in humans determined the level of H2O2, a very unstable substrate. However, catalase was found to be increased in the gastrocnemius of mice upon prolonged exercise [53]; instead, GSH was always affected in all muscle types and increased levels of the tripeptide characterized trained muscle [54]. Recently, a metabolome approach was used in order to characterize the effects of exercise on human skeletal muscle. The results showed that vigorous exercise leads to GSSG and RSSG production as a consequence of increased oxidative stress [55].Behind the detrimental role of excessive ROS, recent evidence has documented that low ROS levels produced during exercise have positive effects by influencing cellular adaptation [
56]. Protein S-glutathionylation represents a reversible and ubiquitous process important for avoiding excessive oxidation of thiol-derivatives of protein cysteine under oxidative stress. The glutathionylation process is mediated by Trx or other “redoxins” and is responsible for protein structure/function changes, therefore, resulting in the modulation of several cellular metabolic processes [
4]. Heart and skeletal muscle contraction and metabolism are regulated by such process through modulation of several ions pumps and contractile proteins [
57,
58]. In fact, mammalian fast-twitch (i.e., type II) muscle fibers exposed to GSH and reactant to induce S-glutathionylation showed an increase in their sensitivity to Ca
2+, which was abrogated by the reducing agent dithiothreitol (DTT), indicating not only modification of protein thiols, but also that glutathionylation of skeletal muscle proteins can modulate Ca
2+ sensitivity [
59]. Moreover, the authors reported that the protein responsible for such an effect was the fast troponin I, which can be S-glutathionylated on Cys134. The most intriguing evidence concerning this protein is that the same Cys residue can be also S-nitrosylated with a concomitant decrease in Ca
2+ sensitivity. This study provides unambiguous evidence on the cross-talk between GSH and NO in muscle functionality: NO counterbalances the ability of oxidant-induced S-glutathionylation to increase Ca
2+ sensitivity, a condition that if persistent would adversely affect skeletal mass performance [
60]. Skeletal muscle mitochondria subjected to in vitro treatment with compounds inducing S-glutathionylation (diamide or disulfiram) produced less ROS as a by-product of aerobic oxidation of either carbohydrates or fatty acid derivatives. This inhibition was due to impaired pyruvate uptake by deactivation of mitochondrial pyruvate carrier and S-glutathionylation of Complex I [
61]. This regulatory process is fundamental in avoiding increased ROS burst that can result in irreversible protein oxidation and mitochondrial damage. The GSH mediated reversible oxidation of specific proteins directly related to ROS production could have a twofold benefit: on one hand, it lowers the harmful effects of high ROS flux and on the other hand, it allows the restoration of protein functions once the intracellular GSH content is reestablished.
As before mentioned also NO has a key role in skeletal muscle as it is involved in several functions including contractility and blood flow. Straightforward evidence of NO requirement in muscle physiology was obtained from experiments with NOSμ-/- animals, in whiche alterations in mitochondrial morphology, bioenergetics and unfolded protein response were demonstrated [
62]. These modifications produced skeletal muscle dysregulation of growth and exercise performance. Generally, NO inhibits force production and modulate Ca
2+ release by direct interaction with sGC and ryanodine receptors, respectively [
63,
64,
65]. Moreover, it inhibits the activity of Ca
2+-dependent sarcoplasmic reticulum (SR) ATPase to avoid the depletion of SR Ca
2+ [
66]. NO avidity to react with heme, the place where also oxygen binds, makes these two gaseous molecules in competition especially into the mitochondria, where high content of heme proteins and Fe-S groups are present to reduce the molecular oxygen concomitantly with ATP production. Indeed, the most recognized effect of NO on skeletal muscle metabolism is its capacity to directly inhibit mitochondrial electron transport chain complexes: NO inhibits electron transfer and NADH-dehydrogenase function at the level of Complex I through the intra-mitochondrial production of ONOO
·− [
67]; NO inhibits electron transfer at Complex III independently of oxygen concentration by inhibiting the transfer of electrons to cytochrome
c and increasing the production of O
2− [
68]; NO inhibits cytochrome
c oxidase activity in dependence of oxygen concentration and at the same time NO can be a good substrate for the enzyme. Then cytochrome
c oxidase engagement with NO can be inhibitory of cellular respiration or for removal of NO from the cell [
67,
69]. At cellular level NO promotes muscle homeostasis by stimulating glucose transport during exercise trough activation of upstream signaling events leading to the translocation of glucose transporter GLUT4 at the cell surface [
70]. One of the demonstrated mechanisms relies on the increase of cGMP level concomitantly with the activation of sGC during exercise. Chemical inhibition of NOS or nNOS knockout in mice muscle abolished such effects while NO donors raise cGMP level and glucose uptake [
71].
NO modulates mitochondrial biogenesis through different routes, a selective one is by increasing the phosphorylated-active form of AMP-activated protein kinase (AMPK) that in turn induces PGC-1a signaling pathway [
72]. PGC-1a is a nuclear and mitochondrial transcriptional coactivator inducing the expression of a large set of genes in response to metabolic and physiological stimuli, such as physical exercise [
73,
74]. Another pathway is shared with ROS by the activation of NFE2L2 expression, which is the master regulator of antioxidant response upon oxidative burst and mitochondrial biogenesis upon exercise in muscle [
75]. NO modulates mitochondrial biogenesis also during myocytes differentiation by S-nitrosylation-mediated activation of the transcription factor CREB (cAMP response element-binding protein), which efficiently binds to the PGC-1a gene promoter region increasing its concentration and downstream signaling events [
76]. Finally, NO produced by the endothelium affects skeletal muscle functions besides its canonical role in the regulation of vascular tone [
77]. In fact, eNOS knockout mice (eNOS-/-) showed a reduction of mitochondrial complexes I and ATP synthase and altered skill in physical exercise: glucose and long-chain fatty acid uptake and glycogenolysis were markedly accelerated [
78].
Through the formation of ONOO
·− or ONOO
·−-derived radical (NO
2), NO is indirectly responsible for irreversible damage to protein residues such as tyrosine nitration. This modification can result in dramatic changes in the target protein structure/function with loss of the activity or acquisition of additional abnormal roles. Moreover, phosphorylative cascades can be altered being tyrosine a residue directly involved in phospho-mediated signaling pathways and the generation of antibodies against nitrated proteins induces immunological responses [
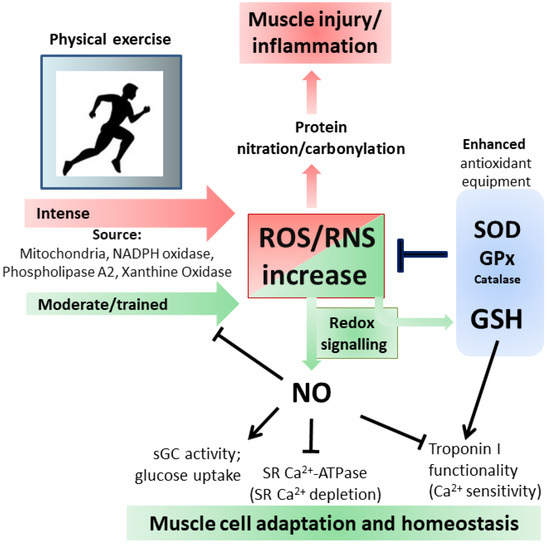
79]. Protein carbonylation, which occurs on arginine, lysine, proline and threonine, represents the most widely studied marker for oxidative damage to proteins by high ROS levels; both carbonylated and nitrated proteins are accumulated in different types of muscle during aging or related pathologies [
80,
81,
82] (
Figure 3).
Figure 3. Oxidative and Nitrosative stress/signaling during intense and moderate physical exercise. For details see the text.
Considering the importance of GSH and NO in the control of muscle responsiveness, nutritional approaches aimed at increasing their concentrations have been tested to preserve muscle homeostasis. For instance, long-term supplementation with a cystine-based antioxidant compound is able to delay age-associated muscle loss [
83]. Direct supplementation of GSH precursors, including N-acetylcysteine, L-alanine and L-glutamine, was also shown to promote muscle performance and recovery during exercise [
84,
85,
86]. Nevertheless, the intake of antioxidants such as Vitamin C and E impedes a number of physiological responses activated during muscle training with no effect on GSH production [
87,
88]. This suggests that the promotion of GSH metabolism rather than blunting ROS production alone is the way for assuring adaptations to exercise [
89]. On the other side, nutritional supplementation of the NO precursors L-Arginine and L-Citrulline has been clearly shown to ameliorate muscle homeostasis in terms of improved protein synthesis rates, myotube diameter and muscle antioxidant capacity [
90,
91]. These phenotypical advantages were also ascribed to the induction of iNOS expression and appreciable in wasting conditions obtained by deprivation of growth factors and/or nutrients.