Ras proteins are essential mediators of a multitude of cellular processes, and its deregulation is frequently associated with cancer appearance, progression, and metastasis. Ras-driven cancers are usually aggressive and difficult to treat. Although the recent Food and Drug Administration (FDA) approval of the first Ras G12C inhibitor is an important milestone, only a small percentage of patients will benefit from it.
- Ras oncogenes
- PI3-Kinase
1. Introduction
Ras proteins are the founding members of the Ras superfamily of GTPases, which in humans is composed of more than 150 members [1,2]. Ras proteins are membrane-bound small GTPases that act as molecular transducers, coupling cell surface receptors to intracellular effector pathways to regulate cellular processes such as cell proliferation, differentiation, migration, and apoptosis [3,4]. In humans, three Ras genes ( H-ras , N-ras, and K-ras ) encode four distinct Ras proteins: H-Ras, N-Ras, K-Ras4A, and K-RasS4B, the latter 2 resulting from alternative RNA splicing of the K-ras gen. These four Ras isoforms are ubiquitously expressed and are highly similar in primary sequence, structure, and biochemical properties [5,6]. They share 90% sequence identity in the G domain [7], being 100% identical in the N-terminus of this domain, termed the effector lobe, and sharing 82% of the sequence of the allosteric lobe [5,8]. In contrast, the C-terminal hypervariable region (HVR) shares little sequence similarity [7,9].
Alterations in Ras signaling are implicated in the development of different diseases, such as neurological disorders, developmental disorders, and autism. Additionally, Ras proteins are recognized as major oncogenes, as mutations in all three Ras genes occur in approximately 30% of human cancers [10]. Mutant Ras is a driver both in tumor initiation and tumor maintenance [5,7]. In general, K-Ras is the most frequently mutated isoform (accounting for 75% of Ras mutation in cancer), followed by N-Ras (17%) and H-Ras (7%) [10]. The frequency in which every isoform appears mutated varies by tissue type: a large percentage of adenocarcinoma of the lung (32%), pancreas (86%), and colon (41%) is driven by K-Ras mutations; 29% of melanomas are driven by mutations in N-Ras, while H-Ras mutations appears mutated in 5% of head and neck squamous cell carcinoma and 6% of bladder cancers [11].
2. Ras Activation and Downstream Signaling
Ras signaling is activated by cellular receptors including receptor tyrosine kinases (RTKs), G-protein coupled receptors (GPCRs), cytokines receptors, and extracellular matrix receptors [12]. Ras proteins act as molecular switches that cycle between two conformational states: an active GTP-bound state and an inactive GDP-bound state. This GDP/GTP cycling is tightly controlled by two main classes of regulatory proteins: guanine-nucleotide-exchange factors (GEFs), which upon receptor stimulation promote the activation of Ras proteins by stimulating GDP for GTP exchange, and GTPase-activating proteins (GAPs), which stimulate Ras-mediated GTP hydrolysis and Ras inactivation [6,12,13]. GTP binding to Ras induces changes in conformation, mainly in two regions named switch I and switch II, that greatly increase the affinity of Ras for its downstream effectors [14,15].
Ras-GTP stimulates a wide range of downstream effectors, although the best characterized are mitogen-activated protein kinases (MAPK), phosphoinositide-3 kinase (PI3K) [16,17], and the Ral pathways [18] ( Figure 1 ). The Raf–MEK –ERK signaling axis was the first Ras-effector that was described [19,20] and it regulates cell growth, differentiation, inflammation, and apoptosis [21]. Activated Ras promotes the translocation of the Raf serine/threonine kinase to the plasma membrane, where it is activated and phosphorylated by different protein kinases. Active Raf phosphorylates and activates the MEK1/2 kinase, which in turn phosphorylates ERK1/2 mitogen-activated protein kinase that eventually exert their function on a large number of downstream molecules [22]. The phosphatidylinositol 3 kinase (PI3K) pathway is the second best-characterized Ras effector and participates in the regulation of a wide range of cellular activities, including cell growth, proliferation, differentiation, migration, and apoptosis [23]. Genetic alterations in PI3K/AKT and Raf/MAPK/ERK pathway components leading to aberrant activation of signaling are frequent in cancer [21,24]. The functional importance of the Ras–PI3K pathway and its role in cancer will be extensively discussed in the next sections.
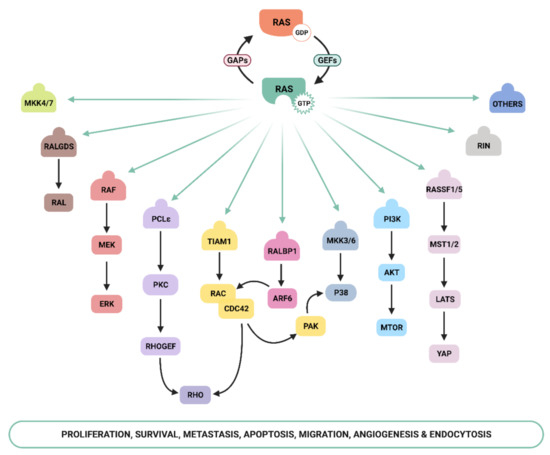
Apart from the above-mentioned effectors, an increasing number of molecules that specifically interact with Ras have been described, such as Ral-GEF, Rho-GTPases , Novel Ras effector 1A (NORE1A), Af6, phospholipase C (PLC), Ras and Rab interactor 1 (RIN1), T-cell lymphoma invasion and metastasis-inducing protein (Tiam), and growth factor receptor 14 (Grb1), although the precise physiological role is still not fully understood for many of them [25]. Recently, Ras signaling has been shown to interact with the Hippo-signaling effector YAP to facilitate tumorigeneses [26]. Thus, the list of Ras effectors continues to grow, and more interactors belonging to the family of Ras effectors are expected to be characterized in the next years [14,27,28,29,30].
3. Ras–PI3K Signaling in the Tumor Microenvironment
Cancer cells dramatically alter their local tissue environment (TME). The clinical relevance of Ras mutations is due not only to their impact on cancer cells’ autonomous mechanisms but also to their ability to alter the way tumor cells interact and influence different components of the TME, from the extracellular matrix (ECM) to endothelial cells, cancer-associated fibroblasts (CAFs), and inflammatory/immune cells. Neo-angiogenesis, tumor inflammation, and matrix remodeling are interconnected processes that alter the expression profile of cancer cells, increasing their aggressiveness, altering their expression profile, and eventually contributing to the end-stage of tumorigenesis represented by tumor dissemination [293].
CAFs are one of the most abundant stromal cell types in the tumor microenvironment and contribute to several aspects of the malignant phenotype [366]. The relevance of Ras and PI3K signaling in CAFs is, by far, less understood than in cancer cells, but different studies have validated the role played by PI3K in the protumoral functions of CAFs. For example, CAF-derived TGF-α promotes the metastatic colonization of ovarian cancer cells via activation of AKT, epidermal growth factor receptor (EGFR), and extracellular signal-regulated kinase (ERK)-1/2 signaling pathways [367]. Activation of PI3K due to loss of PTEN in stromal fibroblasts accelerated initiation, progression, and malignant transformation of mammary epithelial tumors due to the induction of genes involved in ECM remodeling and the recruitment of macrophages, including Mmp9 and Ccl3 [368]. PI3Kγ has been shown to regulate TNF-mediated secretion of matrix metalloproteinases (MMPs) from fibroblasts, which is crucial for the migration of tumor cells [369]. Ziqian Li et al. reported that in colorectal carcinoma and melanoma, CXCL5 derived from the CAFs activated PI3K/AKT signaling pathway and promoted the expression of PD-L1 in tumor cells [370].
Modulation of cancer-associated fibroblasts by K-Ras mutational changes through Hedgehog signaling (Hh) has been suggested as an important mediator of CAF activation in PDAC models [373,374,375,376,377]. SHh originated from cancer cells was responsible for changes on the fibroblast proteome, promoting the synthesis of ECM components, such as collagen, and matrix metalloproteinases (MMPs) [376]. Cancer-derived Hh also promoted the expression of growth factors such as IGF1 and GAS6 by fibroblasts, which reflected back on cancer cells. Additionally, fibroblast-derived signals significantly alter the phosphoproteome of PDAC cells, not only promoting an increase in the usual cell-autonomous alterations but also activating pathways that are not autonomously triggered. Recently, it was shown that the conditioned medium of K-RasV12-overexpressing colorectal cells strongly increased the migration of intestinal fibroblasts without affecting their proliferation rate and their differentiation status [378]. Together, these studies suggest that oncogenic Ras in cancer cells can alter the behavior of fibroblasts, which in turn affects the microenvironment through ECM changes and growth factor signaling, contributing to tumor progression.
It is now evident that Ras signaling not only exerts its activity to drive cell growth and sustain survival in cancer cells, but also modulates microenvironmental changes. In stromal cells, Ras signaling contributes to directing the response to a multitude of cues that cancer cells produce to modify their microenvironment. It is, thus, an important area to explore, not only from basic knowledge, but also from a therapeutic point of view.
4. Targeting Ras and PI3K in Cancer
Hyperactivation of the PI3K pathway in cancer, and its vital function in cell survival and proliferation, have made it an ideal target for treatment. Furthermore, the inhibition of Ras-effector pathways has for years been considered the most effective approach to target Ras-driven tumors [402,403]. Results from clinical trials with PI3K inhibitors in solid tumors have been, however, largely disappointing [404]. Reasons for this failure include drug resistance, aberrant activation of parallel signaling pathways that in turn stimulate the PI3K pathway, and intolerable toxicity due to lack of specificity, especially with pan-PI3K inhibitors [74,405], while isoform-specific inhibitors seem to have better therapeutic efficacy and an improved toxicity profile [406]. So far, only five PI3K inhibitors have been approved to use in clinical practice. One of them is Alpelisib, a PI3Kα inhibitor approved for breast cancer treatment and investigated in combination with a range of targeted therapies and chemotherapies in multiple clinical trials [407]. The other PI3K inhibitors are used in the treatment of hematological cancers: idealisib, a PI3Kδ inhibitors [404], copanslisib, the only pan-PI3K inhibitor approved [408], duvelisib, a dual inhibitor of the PI3Kδ and PI3Kγ isoforms [409], and umbralisib, a dual inhibitor of PI3Kδ/casein kinase-1εr ( www.fda.gov , (accessed on 20 May 2021) [410]).
The emerging roles of Ras and PI3K in shaping the tumor microenvironment provide a new awareness of how kinase inhibitors may contribute to enhancing antitumor immunity and their application in combination therapy to improve the outcome of immunotherapy. For example, PI3Kα or PI3Kβ inhibitors impair tumor cell growth and induce changes in the tumor microvasculature, which correlates with antitumor activity and, more importantly, potentiates the anticancer activity of current cytotoxic therapy [319,321,414,415]. Furthermore, PI3Kβ inhibition could improve the efficacy of immunotherapy by increasing T-cell infiltration in melanoma with PTEN loss [416]. Additionally, in leukemic B-cells, PI3Kδ blockade impairs pro-survival signaling and induces tumor cell death. On the other hand, the inhibition of PI3Kδ in monocyte-derived cells of the lymph node stroma blocks the secretion of pro-survival chemokines sensed by tumor cells, potentiating the pro-apoptotic effect of the drug [417].
The same principle could be applied to PI3Kγ and PI3Kδ specific inhibitors used for the treatment of solid tumors. PI3Kδ inactivation protects against the development of a broad range of cancers, which is mainly associated with the disruption of the function of Tregs and possibly of MDSCs [351]. The anticancer activity of PI3Kγ seems to be associated with its effects on tumor macrophages, which translates into the stimulation of T-cell recruitment and reduced tumor growth [335]. Important results with PI3Kδ and PI3Kγ inhibitors, used alone or in combination with standard anticancer therapy, have been obtained in murine models of pancreatic cancer, a tumor that is highly resistant to chemotherapy and radiation therapy and does not respond to immune checkpoint inhibitors [98,335,351,418,419]. For example, the deletion of PI3Kγ improved survival and responsiveness to standard-of-care chemotherapy in animal models of PDAC [335], and the dual inhibition of PI3Kγ and CSF-1/CSF-1R reduced the tumor infiltration of MDSCs and inhibited tumor growth [420]. Importantly, PI3Kγ inhibition can synergize with anti-PD-1 to reduce tumor growth, extending survival [421,422], and pan-PI3K inhibition can be used to overcome resistance to immune checkpoint inhibitors [423].
This entry is adapted from the peer-reviewed paper 10.3390/genes12071094