The functional neural circuits are partially repaired after an ischemic stroke in the central nervous system (CNS). In the CNS, neurovascular units, including neurons, endothelial cells, astrocytes, pericytes, microglia, and oligodendrocytes maintain homeostasis; however, these cellular networks are damaged after an ischemic stroke. Commonly shared molecular mechanisms in the neurovascular unit are associated with the vascular endothelial growth factor (VEGF) and its related factors. Stem cells and gaseous molecules may exert therapeutic effects by diminishing VEGF-mediated vascular leakage and facilitating VEGF-mediated regenerative capacity.
1. Overview of Repair Mechanisms Following Brain Damage
Ischemic stroke, which accounts for 87% of all stroke cases, results from a sudden cessation of adequate amounts of blood supply to parts of the brain. The vascular system plays a critical role in supplying oxygen (O2) and nutrients to neuronal systems. An ischemic stroke typically presents with a rapid onset of neurological deficit. Cell–cell communication in the neurovascular unit contributes to a functional neurovascular system through an orchestrated network of extracellular matrix, endothelial cells (ECs), pericytes, astrocytes, oligodendrocytes, microglia, neural stem cells, and neurons. Thus, interruption of blood flow through an intracranial artery leads to deprivation of O2 and nutrients to the vascular territory, resulting in metabolic changes in the surrounding cells, such as abnormal mitochondrial activity, inflammation, disruption of the blood–brain barrier (BBB), and cell death. Functional recovery after an ischemic stroke may depend on the fate of the ischemic penumbra if the circulation is re-established in time. If not, at the onset of a stroke, the complex and dynamic association between the brain vasculature and neuronal system delays functional recovery.
Elucidating the mechanism of neurovascular repair is important to develop therapeutic strategies to attempt to reverse or minimize the effects and to prevent future infarcts. Clinical trials aiming to develop strategies for neuroprotection with respect to ischemic stroke have failed to demonstrate clinical efficacy, possibly due to limited regenerative capacity in the central nervous system (CNS) [
1]. In this review, we specifically discuss the repair mechanisms related to neurovascular protection and regeneration through stem cell-mediated repair mechanisms and gaseous molecule-mediated regenerative signaling. Additionally, strategies for overcoming a stroke are also discussed, with a specific focus on cellular therapy and molecular mechanisms involving vascular endothelial growth factor (VEGF). The VEGF family includes VEGF-A, -B, -C, -D, -E, and placental growth factor, which bind in a distinct pattern to three structurally related receptor-type tyrosine kinases, namely, VEGF receptor 1 (VEGFR1), VEGFR2, and VEGFR3. VEGF-A is a major growth factor that binds to VEGFR1 and VEGFR2 but not to VEGFR3 [
2].
2. Neurovascular Repair
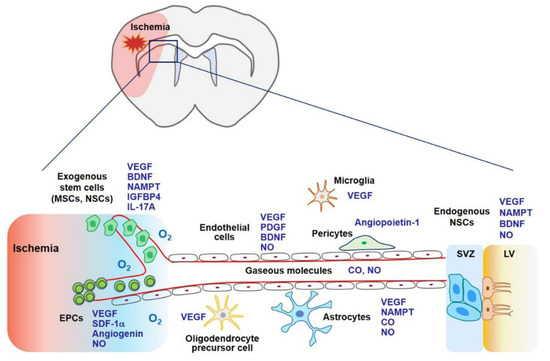
Stem cell therapy using MSCs, endothelial progenitor cells (EPCs), and neural stem cells (NSCs) may contribute to stroke recovery as stem cells can secrete a variety of cytokines and growth factors related to angiogenesis, neurogenesis, and synaptogenesis (
Figure 1) [
93]. Gaseous molecules such as NO and CO also possess regenerative potential demonstrated by boosting stem cell-mediated repair or by stimulating additional regenerative pathways [
58,
94,
95]. Both pathways share common molecular mechanisms associated with VEGF and its related factors (
Figure 1).
Figure 1. In the later phase of an ischemic stroke, VEGF expression may facilitate tissue regeneration. Exogenous stem cells (i.e., MSCs, EPCs, and NSCs) and gaseous molecules (i.e., NO and CO) increase the expression of VEGF and its related factors, which are involved in angiogenesis, neurogenesis, and synaptogenesis. Additionally, these factors enhance the ability of endogenous NSCs to proliferate and differentiate into mature neurons, astrocytes, and oligodendrocytes. Collectively, regeneration through stem cells and gaseous molecules contributes to repair after an ischemic stroke. Abbreviations: MSCs, mesenchymal stem cells; NSCs, neural stem cells; EPCs, endothelial precursor cells; BDNF, brain-derived neurotrophic factor; IGFBP4, insulin-like growth factor binding protein-4; PDGF, platelet-derived growth factor; SDF-1α, stromal cell-derived factor-1α; SVZ, subventricular zone; LV, lateral ventricle.
2.1. Stem Cell Therapy
Progress in stem cell biology has significantly contributed to the development of strategies for the treatment of strokes in preclinical studies and has demonstrated clinical potential in stroke treatment. Stem cell therapy may be a promising strategy for the treatment of intractable neurological diseases in the future. Transplanted exogenous stem cell therapy for a brain ischemic stroke may contribute directly to neurovascular regeneration (by compensating for the loss of nerve tissue by the differentiation of nerve and glial cells) as well as indirectly (via secretion of angiogenic and neurogenic factors from the penumbra brain regions). Here, we discuss MSCs, EPCs, and NSCs as potential therapeutic cell types that might be beneficial for the treatment of strokes.
2.1.1. Mesenchymal Stem Cells
MSCs can differentiate into chondrocytes, adipocytes, and osteoblasts, as well as transdifferentiate into ECs, glial cells, and neurons. Owing to their remarkable regeneration potential, MSCs are widely used in current medical research [
96]. MSCs secrete a wide range of growth factors, cytokines, chemokines, and extracellular vesicles, thereby contributing to the repair process (i.e., angiogenesis, gliogenesis, and neurogenesis) after an ischemic stroke [
97].
The therapeutic potential of MSCs has been demonstrated in ischemic animal models. Human MSCs have been shown to enhance stroke lesion recovery by mediating inflammation and tissue repair through the secretion of trophic factors. Human MSC transplantation into a rat focal ischemia model of transient cerebral artery occlusion revealed decreased accumulation of Iba-1-positive microglia and GFAP-positive astrocytes and the inhibition of pro-inflammatory gene expression in the core and ischemic border zone [
87]. MSC therapy may improve outcomes of an ischemic stroke by inhibiting the activity of the proinflammatory phenotype of microglia but augmenting the activity of the anti-inflammatory phenotype of microglia [
85]. Human umbilical cord blood-derived MSCs (intravenous injection, 0.25 million cells/animal and 1 million cells/animal) were injected into a rat ischemia–reperfusion stroke model, and experiments were conducted 7 days after reperfusion [
98]. The treatment reduced the mRNA and protein levels of metalloproteinases (MMPs) (i.e., MMP-9 and MMP-12) [
98].
Apart from the anti-inflammatory responses, MSCs stimulate the regenerative pathway. B10 human MSCs express cytokines and growth factors, including IL-5, fractalkine, insulin-like growth factor-1, glia-derived neurotrophic factor, and VEGF [
87]. B10 transplantation also increases the expression of angiogenic factors, such as HIF-1α in the core and border zone of rat ischemic stroke brains [
99], which can induce VEGF expression and new vessel formation [
99,
100]. Allogeneic adipose-derived MSC sheets demonstrated neurological improvement with angiogenesis and neurogenesis in a rat stroke model [
101]. Mitochondrial transfer from MSCs to cerebral microvasculature resulted in significant improvement of the mitochondrial activity in injured microvasculature, enhanced angiogenesis, reduced infarct volume, and improved functional recovery following an ischemic stroke [
102].
Stromal cell-derived factor-1α (SDF-1α)-transfected MSCs enhance ischemia-mediated new vessel formation as well as angiogenesis in vivo via the VEGF–eNOS axis [
103]. IFN-γ-activated MSCs were injected into a rat MCAO model. IFN-γ-activated MSCs demonstrated more potent functional recovery as assessed by the modified neurological severity score and open-field analysis compared to that observed in vehicle-treated animals [
104]. IFN-γ-activated MSC-treated stroke-conditioned animals showed a reduction in infarct size, diminished microglial activation, and enhanced recruitment and differentiation of OPCs to myelin-producing oligodendrocytes [
104]. Intra-arterial transplantation of 3-dimension (3D) aggregate-derived human MSCs into transient MCAO stroke model mice exhibited increased cell persistence and better therapeutic outcomes compared to that in saline control or 2D human MSC control [
105]. The PI3K–Akt signaling pathway was activated by 3D-aggregate-human MSCs [
105]. The extracellular regulating kinase 1/2 (ERK) pathway is considered an important regulator in CNS regeneration [
58,
106]. ERK-overexpressing MSCs were transplanted into stroke model rats, demonstrating the increased proliferation of NSCs and maturation into neurons in the subventricular zone [
107]. Glia-like human MSCs (ghMSCs) exhibit better efficacy and enable better protection of the neurons and the brain from ischemia than naïve human MSCs, and insulin-like growth factor binding protein-4 (IGFBP-4) played a critical role in mediating the beneficial effects of ghMSCs in an ischemic stroke [
108]. IGFBP-4, hepatocyte growth factor, and VEGF released from ghMSCs may serve as key molecules for enhanced neuronal survival and neurite outgrowth in ischemic CNS injuries [
108,
109]. Small extracellular vesicles secreted by human-induced pluripotent stem cell-derived MSCs enhance angiogenesis by inhibiting STAT3-dependent autophagy in a rat model of an ischemic stroke [
110]. MSC transplantation has also been investigated in humans. Autologous MSC transplantation (intravenous injection, 1 × 10
8 cells) may improve neurological functions one year after symptom onset in stroke patients [
111]. In this study, of the 31 enrolled patients, 16 were administered MSCs. The MSC-treated group showed improvements in motor functioning based on the examination of the National Institutes of Health Stroke Scale score and Fugl-Meyer scores as well as in task-related functional magnetic resonance imaging activity [
112]. The transplantation of autologous human MSCs (intravenous injection), cultured in human serum, was performed in 12 stroke patients [
113]. In this unblinded study, the mean lesion volume, as assessed by magnetic resonance imaging, was reduced by 420% at one week post-cell infusion [
113]. Allogeneic ischemia-tolerant MSCs (intravenous injection, 0.5, 1.0, and 1.5 million cells/kg body weight) were transfused into patients with chronic stroke. Their Barthel index scores increased at 6 months and 12 months post-infusion [
114]. Taken together, MSCs may exhibit therapeutic potential by inhibiting excessive inflammation and stimulating the repair pathway.
2.1.2. Endothelial Progenitor Cells (EPCs)
The formation of new blood vessels in the adult brain after a stroke stems from angiogenesis (migration and proliferation of local mature ECs) and the systemic regulation of bone marrow-derived EPCs [
93]. CD34 and VEGFR2 double-positive mononuclear cells from peripheral blood are considered as EPCs [
115]. EPC mobilization from the bone marrow stroma into the blood circulation is regulated by various enzymes and factors such as eNOS, VEGF, and granulocyte colony-stimulating factor (G-CSF) [
43,
116,
117]. The SDF-1α/C-X-C motif chemokine receptor 4 (CXCR4) pathway plays a key role in the homing of EPCs to the ischemic region [
93,
118]. The SDF-1α–CXCR4 interaction may recruit not only EPCs, but also MSCs and NSCs to ischemic tissues since the SDF-1–CXCR4 axis modulates survival, proliferation, migration, and differentiation of MSCs and NSCs [
119,
120].
Ex vivo expanded EPCs (intravenous injection, 1 × 10
6 cells) were injected into mice after 1 h following induction of transient MCAO [
121]. EPC transplantation significantly reduced ischemic infarct volume and induced angiogenesis in the ischemic penumbra after MCAO compared to that observed in control mice in vivo, and a CXCR4 antagonist blocked SDF-1-mediated EPC migration in vitro [
121]. Moreover, SDF-1 upregulates VEGF expression and eNOS activity via cellular communication [
103].
The interplay between eNOS and BDNF may be involved in EPC-mediated angiogenesis, neurogenesis, and axonal growth after an ischemic stroke [
122]. Conditioned media derived from EPC culture was administered to mice 1 d after MCAO. A significant increase in capillary density was observed in the ischemic penumbra, consequently improving forelimb strength [
123]. The expression of multiple growth factors, cytokines, and proteases has been demonstrated in the EPC secretome, showing enhanced endothelial and OPC proliferation and maturation [
90]. Angiogenin, a HIF-1α target gene, may be a key factor since pharmacological blockade of angiogenin signaling negates the positive effects of the EPC secretome [
90,
124]. Under in vivo conditions, treatment with the EPC secretome increases vascular density, myelin, and mature oligodendrocytes in the white matter and rescues cognitive function in a mouse model of hypoperfusion [
90]. Hypoxic preconditioning via overexpression of HIF-1α, SDF-1α, VEGFR2, or VEGF may facilitate EPC functions such as angiogenesis and neurogenesis [
125,
126].
Autologous CD34-positive stem/progenitor cells derived from the bone marrow of human subjects were administered intra-arterially via catheter angiography within 7 days of the onset of a severe ischemic stroke [
127]. In this study, administration of CD34-positive stem cells resulted in reduced lesion volume and hence, rescued patients with an acute ischemic stroke during a 6-month follow-up period [
127]. Thus, the application of EPCs in an ischemic stroke may be helpful.
2.1.3. Neural Stem Cells (NSCs)
IL-17A shows two distinct peaks of expression in the ischemic hemisphere: the first peak observed within 3 days and the second on day 28 after a stroke. Astrocytes are a major cellular source of IL-17A, which maintains and augments subventricular zone (SVZ) neuronal precursor cell survival, neuronal differentiation, synaptogenesis, and functional recovery after a stroke [
128]. In this study, the p38 mitogen-activated protein kinase–calpain 1 signaling pathway was involved in IL-17A-mediated neurogenesis [
128].
NAMPT is a rate-limiting enzyme involved in the biosynthesis of nicotinamide adenine dinucleotide (NAD) in mammals; this putative therapeutic agent for combating stroke is highly expressed in neurons, EPCs, and NSCs, with lower expression in glial cells [
129,
130]. It plays key roles in defense mechanisms, metabolic homeostasis, and neuronal survival [
129]. NAD replenishment in neurons either before or after oxygen-glucose deprivation reduces cell death and DNA damage [
131]. Neuronal survival due to NAMPT overexpression was blocked in AMPKα2−/− neurons through the SIRT1–AMPK axis in a rat model of an ischemic stroke [
132]. The role of NAMPT has been demonstrated in neurovascular repair during the chronic phase. NAMPT promotes angiogenesis, neovascularization, and neurite outgrowth as well as increases the levels of regenerative factors such as BDNF and VEGF [
129,
133,
134,
135].
2.2. Gaseous Biomolecules
Reduced O
2 availability (i.e., hypoxia) and energy substrates (e.g., glucose) appear to represent the critical stimulus that evokes an adaptive response to ischemia, principally through HIF-dependent production of multiple angiogenic cytokines and growth factors, including VEGF, angiogenin, angiopoietins, placental growth factor, PDGF-B, stem cell factor, and SDF-1, which stimulate angiogenesis, the process of new blood vessel formation from pre-existing ones [
27,
124,
136,
137,
138,
139].
The endogenous repair ability of gaseous biomolecules such as NO and CO can be promoted after a stroke. Nitric oxide (NO) and CO are endogenous gases produced by NOS and HO, which diffuse freely between cells, consequently amplifying signaling pathways involved in neurogenic and angiogenic functions [
42,
137]. NO can dilute the cerebral vasculature and enhance cerebral blood flow. CO improves damaged vasculature by inducing angiogenesis and neovascularization, partly by interacting with the NOS signaling pathway [
94]. Moderate levels of NO and CO induces HIF-1α-mediated VEGF expression [
75,
140] and suppress its expression in severe hypoxia [
141].
3.2.1. NO
NO is produced by the reaction of l-arginine with NOS isoforms. Two constitutive isoforms, (eNOS and neuronal NOS [nNOS]) via Ca
2+ entry, and iNOS are enzymes that are expressed in a highly cell type-specific manner. Acting as an intercellular signal, the nNOS–NO axis can trigger neurogenesis in mouse brain neural progenitor cells. BDNF upregulates nNOS protein levels, which can induce the maturation of neurons from neural progenitor cells [
142].
VEGF–VEGFR2–PI3K–Akt axis is an important regulator of cellular survival, cell motility, and NO production [
143,
144]. Activation of AMPK, a crucial cellular energy sensor, can also stimulate eNOS by phosphorylating it at Ser
1179, suggesting crosstalk between cellular metabolism and vascular tone. By using flow channels with cultured ECs, AMPKα Thr
172 phosphorylation can be increased with changes in flow rate or pulsatility [
145].
eNOS exhibits neuroprotective properties against ischemic strokes [
43]. Moderate NO gas inhalation in mice with transient focal ischemia reduced infarct volume to 10 ppm for 24 h, and to 20, 40, and 60 ppm for 8 and 16 h following NO inhalation [
146]. NO inhalation improves penumbral blood flow and neurological outcomes in a mice ischemia induced by transient MCAO [
147] and in a rat model of focal cerebral ischemia [
63].
An HO inducer or carbon monoxide-releasing molecule 2 (CORM-2) restores TNF-α-induced downregulation of the expression of eNOS–NO by inhibiting NF-κB-responsive miR-155-5p expression in HUVECs [
42]. CO can reduce the production of ROS, consequently reducing the synthesis of peroxynitrite [
148]. Application of CORM-2 in BV2 microglial cells prevents the production of NO upon lipopolysaccharide (LPS) stimulation [
149]. Therefore, CO may reduce LPS-mediated NO production in activated glial cells and stimulate NO production in vascular cells. In addition, CO may facilitate repair and regeneration by activating the nNOS–NO pathway in neuronal cells [
95]. The interplay between CO and NO leads to vascular dilation, angiogenesis, and neurogenesis.
The nNOS–NO axis is activated in the neurons and NSCs when mice are injected with CORM-3 after an ischemic CNS injury [
95]. CO may exert neurogenic effects by stimulating the HO-1 pathway, consequently activating the nNOS–NO axis. The HO metabolite, bilirubin, stimulates ERK1/2 phosphorylation, CREB phosphorylation, and nNOS–NO production in the absence of exogenous growth factors in PC12 cells. This effect is blocked by an extracellular Ca
2+ chelator [
150]. NMDAR, a critical receptor for hippocampal long-term potentiation and spatial learning, is S-nitrosylated by NO [
150]. Thus, the crosstalk between HO/CO and NOS/NO may induce diverse signaling pathways leading to neurogenesis, long-term potentiation, learning, and memory.
2.2.2. Carbon Monoxide (CO)
HO-1 and HO-2 are essential enzymes in heme catabolism that cleave heme to carbon monoxide (CO), biliverdin (which is rapidly converted to bilirubin), and Fe
2+. In this step, O
2 is required as the reaction substrate [
136]. HO-2 is constitutively expressed in neurons where it functions as an intrinsic protector [
151]. The expression of HO-1 is strongly induced in various cells in response to hypoxia and stress, which promotes neuroprotection and angiogenesis in the ischemic milieu [
152]. CO induces the expression of HO-1 and plays important roles in neurotransmission, neurogenesis, mitochondrial biogenesis, and blood circulation in the brain [
153,
154,
155]. CO generated either by exogenous delivery or by HO activity is fundamentally involved in regulating mitochondria-mediated redox cascades for adaptive gene expression and improving blood circulation (i.e., O
2 delivery) via neovascularization, leading to the regulation of mitochondrial energy metabolism [
137]. CO can be delivered in a pharmacologically active form as a CO-releasing molecule, CORM [
156]. The biological effects of CO are largely dependent on the HO activity [
137].
HO-derived CO promotes angiogenesis and neovascularization by regulating pro-angiogenic VEGF expression [
157,
158,
159,
160]. CO can also upregulate HO-1 expression, and the HO-1/CO circuit may interact with the NOS/NO pathway [
94]. CORM-2 can stimulate the eNOS–NO axis through inositol triphosphate receptor-mediated intracellular Ca
2+ release, PI3K-Akt phosphorylation, and eNOS dimerization in HUVECs [
161]. CORM-2 prevents TNF-α-induced eNOS downregulation by inhibiting NF-κB-responsive miR-155-5p biogenesis [
42]. Inflammatory responses after ischemic stroke are diminished by CORM-3, demonstrated by examining the levels of TNF-α and IL-1β [
162]. CO-mediated ROS/RNS inhibition may protect the BBB from acute neuroinflammatory diseases [
95,
162].
Signaling pathways activated by CORM-2 include the PI3K-HIF-1α–VEGF pathway [
75]. In a mouse model of ischemia–reperfusion injury, HO-1 is expressed in astrocytes in the penumbra region [
19]. Transient HO-1 activation may be beneficial for regeneration during an acute ischemic injury [
95,
156,
163,
164,
165,
166]. HO-1-derived CO and bilirubin activate LTCC and mediate Ca
2+/CaMKKββ-mediated activation of AMPKα, AMPKα-dependent HO-1 induction, and the consequent stabilization of HIF-1α in a PHD2-dependent manner [
49]. The effects of CO on regeneration are associated with VEGF production [
19]. Recently, the neuroprotective and regenerative effects of CORM-3 have been demonstrated in a stroke. CORM-3 injection reduced infarct volume and increased the expression of mature neuronal markers such as neuronal nuclear antigen and microtubule-associated protein 2 compared to that in saline-treated mice [
162]. CO-mediated VEGF upregulation does not disrupt BBB, instead, CO protects the BBB from ischemic injury [
95,
162]. Other CO-mediated protective factors may mitigate VEGF-mediated vascular permeability, or CO reduces excessive VEGF production. Taken together, CO reduces VEGF-mediated disruption of BBB and facilitates VEGF-induced regeneration after an ischemic stroke.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22168543