Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Genetics & Heredity
Most DFNB1 phenotypes are described as prelingual and bilateral non-syndromic hearing loss, this being severe to profound. This type of deafness affects all frequencies and is not associated with inner ear malformations.
- WGS
- DFNB1
- deafness
- variants
- functional assays
1. Introduction
Hearing loss is the most common sensory pathology, affecting about 1–2 in every 1000 newborns, with a prevalence which increases with age [1,2,3,4]. In industrialized countries, congenital deafness has a genetic origin in 80% of cases [5]. Deafness can be syndromic or not (associated or not with other pathologies or malformations), respectively representing 10% and 90% of cases. More than 500 syndromes are associated with syndromic deafness and more than 100 genes have been described in non-syndromic hearing loss (NSHL) (Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage. https://hereditaryhearingloss.org, the 10 May 2021).
NSHL can be classified according to heredity. Generally 80–90% of those affected have autosomal recessive inheritance (DFNB), 10–15% have a dominant mode (DFNA), 1% of cases are associated with the X chromosome (DFNX), and others have a mitochondrial inheritance mode [5,6].
The predominant form is autosomal recessive non-syndromic hearing loss (DFNB1). Most DFNB1 phenotypes are described as prelingual and bilateral non-syndromic hearing loss, this being severe to profound. This type of deafness affects all frequencies and is not associated with inner ear malformations. Vestibular function remains unaffected [1,2,3,4,5,7,8]. The GJB2 (Gap Junction β 2-chr13:20,187,470–20,192,938 (hg38)) gene is mainly implicated in DFNB1 with frequencies ranging from 20% to 40%, according to populations with the most frequent mutation, c.35delG [3,5,9,10,11].
Moreover, seven large DFNB1 deletions have been described in DFNB1 patients: del-920 kb [12], del-101 kb del(GJB2-D13S175) [13], del(GJB6-D13S1830) [14], del(GJB6-D13S1854) [15], del-131kb [16], del-179kb [17], and del-8kb [18]. This year, Brozkova et al. described a DFNB1 deletion of 3 kb in one patient [11].
The genomic architecture of our chromosomes is now far better investigated and understood. Many studies have focused on the role of non-coding regions and on genetic variants they contain, opening up new research possibilities. However, a vast majority of coding and non-coding variants may remain of unknown clinical significance [19,20,21].
Almost 8% of the human genome is covered with candidate cis-regulatory elements (cCREs) [22]. The identification of distal acting regulatory elements has been the object of active research in recent years. Disruptions of such regulatory elements and/or chromatin conformation are likely to play a critical role in human genetic diseases [19,20,21].
Routine molecular diagnosis in the Molecular Genetics Laboratory at Brest University Hospital involves the testing of around 80 deaf patients each year, and among these patients, ~20% are DFNB1 biallelic carriers. However, several genotypes remain incomplete; for monoallelic DFNB1, which represents fewer than 1% of the tested patients, most patients are carriers of the c.35delG heterozygous and some have rare variants.
The c.35delG heterozygous genotype may be related to the general population frequency with an overall frequency of 2% but an excess of the deletion has been shown in the deaf population [8].
2. GJB2 Mutations
WGS analysis identified a second mutation on the GJB2 gene in patients P3, P4, P8, and P10 (Table 2).
Table 2. DFNB1 mutations detected by WGS.
| Patient | Gene | HGVSc | Chr. | Position (hg38) | HGVSp | Impact | Consequences | Variant Class | rs Number | Allele Frequency (GnomAD) |
|---|---|---|---|---|---|---|---|---|---|---|
| P3 | GJB2 | NM_004004.5:c.269dup | chr13 | 20189312 | NP_003995.2:p.Val91SerfsTer11 | HIGH | frameshift_variant | insertion | rs730880338 | 0.00002940 |
| GJB2 | NM_004004.5:c.269T > C | chr13 | 20189313 | NP_003995.2:p.Leu90Pro | MODERATE | missense_variant | SNV | rs80338945 | 0.001161 | |
| P4 | GJB2 | NM_004004.5:c.35del | chr13 | 20189546 | NP_003995.2:p.Gly12ValfsTer2 | HIGH | frameshift_variant | deletion | rs80338939 | 0.009802 |
| GJB2 | NM_004004.5:c.269dup | chr13 | 20189312 | NP_003995.2:p.Val91SerfsTer11 | HIGH | frameshift_variant | insertion | rs730880338 | 0.00002940 | |
| P8 | GJB2 | NM_004004.5:c.139G > T | chr13 | 20189443 | NP_003995.2:p.Glu47Ter | HIGH | stop_gained | SNV | rs104894398 | 0.0001176 |
| GJB2 | NM_004004.5:c.-23 + 1G > A | chr13 | 20192782 | . | HIGH | splice_donor_variant | SNV | rs80338940 | 0.0003236 | |
| P10 | GJB2 | NM_004004.5:c.139G > T | chr13 | 20189443 | NP_003995.2:p.Glu47Ter | HIGH | stop_gained | SNV | rs104894398 | 0.0001176 |
| GJB2 | G > C | chr13 | 20193022 | - | UNKNOWN | upstream_gene_variant | SNV | rs1425012952 | 0.00001472 | |
| GJB2 | G > T | chr13 | 20183294 | - | UNKNOWN | downstream_gene_variant | SNV | rs372782198 | 0.003099 |
In the routine molecular diagnosis, the Sanger sequencing of the GJB2 gene of patient P3 allowed us to detect frameshift variation (rs730880338) at c.269. Then, a missense variation (rs80338945) was discovered by WGS analysis at the same position. Indeed, these variations in the same nucleotide complicated the interpretation of Sanger sequencing analysis. The duplication hid the other mutation so that the initial Sanger sequencing interpretation failed to detect the two mutations.
DNA samples from parents were not available for segregation analysis, but single-strand NGS sequencing (see IGV (Integrative Genome Viewer) BAM visualization (Figure 1) confirmed that these variants are in trans.
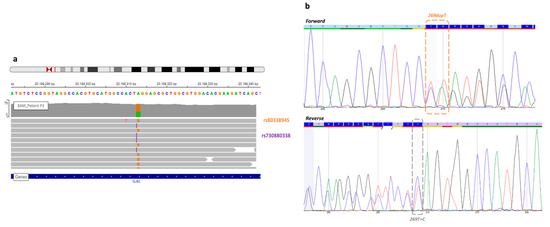
Figure 1. View of the missense variant and frameshift GJB2 gene of patient P3. (a) In the Integrative Genome Viewer, the frameshift variant (rs730880338) at c.269 GJB2 position, known before WGS (orange), and the missense variant (rs80338945) discovered by WGS analysis (purple). Each variant was on a different read, so this analysis confirmed a trans configuration. (b) A new Sanger sequencing in forward and reverse detected both mutations, but it still remains difficult to interpret.
Patient 4 carried two GJB2 mutations, a recurrent mutation, in deaf population, only the c.35delG (rs8033893) have been detected by DHPLC (Denaturing High Performance Liquid Chromatography). The second mutation discovered by WGS analysis was a frameshift (rs730880338), c.269dup (Figure 2). Indeed, since no hetero- and homo-duplex was detected by HPLC we did not realize Sanger sequencing. This is why genotype was unresolved before WGS. Variant segregation of parents allowed the determination of variants transmission for their children (Figure 3).

Figure 2. View of GJB2 variants of patient P4 in IGV. (a) WGS confirmed the known c.35delG. (b) The frameshift variation, c.269dup, detected via WGS and observed in IGV.
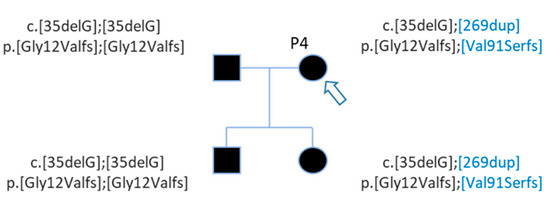
Figure 3. Pedigree of patient P4 with 2 GJB2 variants. Patient P4 (arrow) carries two GJB2 mutations, the c.35delG known before WGS and the c.269dup discovered by WGS analysis (in blue). WGS analysis detected mutations for her daughter also.
The first Sanger sequencing of Patient P8 was performed in 2002; only the nonsense mutation of GJB2 was detected (rs104894398—c.139G > T) (Figure 4).

Figure 4. View of GJB2 variants of patient P8 in IGV. (a) The nonsense variation, c.139G > T, discovered during routine care. (b) The second GJB2 variation detected by WGS analysis is a splice site mutation.
WGS analysis allowed identification of a second GJB2 mutation, a splice site mutation (rs80338940—c.-23 + 1G > A) on intron 1 of the GJB2 gene (Figure 4). This variant had not been found earlier because the sequencing of exon 1 has only been routinely done in the laboratory since 2005.
After WGS, Sanger sequencing confirmed this second mutation, and segregation confirmed that these mutations are in trans (Figure 5). The genotypes of patient P8 and her brother are c.[-23 + 1G > A];[139G > T] p.[?];[Glu47Ter] (Figure 5).
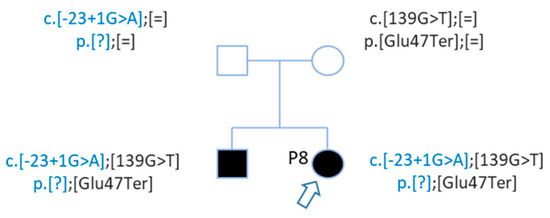
Figure 5. Pedigree of patient P8 with 2 GJB2 mutations in trans. Patient P8 and her brother carried 2 mutations in trans. Mutations were inherited from each parent.
This entry is adapted from the peer-reviewed paper 10.3390/genes12081267
This entry is offline, you can click here to edit this entry!