The tumor suppressor BRCA2 functions as a central caretaker of genome stability, and individuals who carry BRCA2 mutations are predisposed to breast, ovarian, and other cancers.
- homologous recombination
- DNA repair
- fork protection
- genome stability
1. Introduction
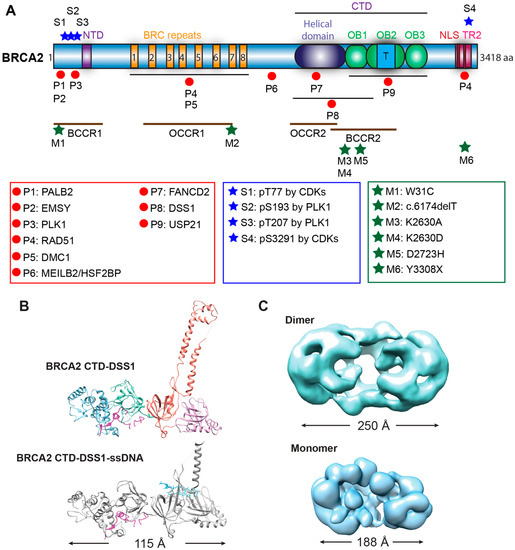
Genome instability is a fundamental cause of tumorigenesis, and the DNA damage response coordinates multiple DNA repair pathways at the frontline guarding genome integrity. BRCA2 is a prototypical caretaker gene, and its inactivation leads to genomic instability that is associated with oncogene activation and loss of tumor suppressor genes [1]. Thus, individuals who carry BRCA2 mutations have elevated risks for breast, ovarian, prostate, pancreatic, and other cancers [2,3]. BRCA2 -deficient cells exhibit severe DNA repair phenotypes, including gross chromosomal rearrangements, accumulation of chromatid breaks, and hypersensitivity to genotoxins [4,5]. BRCA2 plays a conspicuous role as a mediator in homologous recombination (HR), a high-fidelity DNA repair pathway for double-strand breaks (DSB), and interstrand crosslinks (ICL) with additional functions in DNA replication fork support ( Figure 1 ) [6,7]. In the last ten years, mounting evidence has demonstrated a direct role of BRCA2 in replication fork protection (RFP), both in an HR-dependent and HR-independent manner [8]. Recently, BRCA2 has also been shown to suppress other DSB repair pathways and prevent genomic rearrangements, independent of RAD51 [9]. BRCA2 engages in a multitude of protein interactions affecting its function as well as nuclear or chromosomal localization, including DSS1, PALB2, and EMSY (Figure 2) [10,11,12,13].
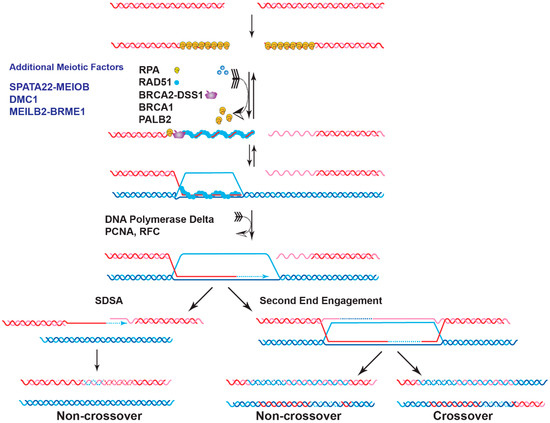
2. BRCA2 as a Mediator of DNA DSB Repair
Deficiency of BRCA2 leads to defective HR repair of chromosomal breaks [16,17]. A primary role of BRCA2 in genome stability maintenance is to facilitate the assembly of the nucleoprotein filament of RAD51, the central enzyme in HR. The RAD51 filament is the catalytic scaffold for homology search and DNA strand invasion, the central steps of HR ( Figure 1 ). The 3′ ssDNA overhang generated by DSB resection sets the stage for the orchestrated turnover between initial occupancy of RPA , the eukaryotic ssDNA-binding protein, and later binding of RAD51 and additional proteins such as the RAD51 paralogs and RAD54 [14]. BRCA2 is required for this timely exchange to enable the formation of RAD51 filaments, as shown by biochemical reconstitution with purified full-length BRCA2 protein and supported by genetic and cell biological analyses [4,5,18,19,20,21,22]. BRCA2 is not known to play a direct role in the ensuing steps of D-loop formation, HR-associated DNA synthesis, and the processing of joint molecules to restore intact chromosomes ( Figure 1 ). This section will review the current status of the structure of BRCA2 and discuss the recent progress in characterizing BRCA2 as a mediator protein focusing on three key biochemical functions: DNA binding, turnover of the RPA -ssDNA complex, and RAD51 loading.
The mechanism of recombination mediator proteins has been highly conserved from bacteria to eukaryotes [52]; E. coli RecFOR and the U. maydis BRCA2 homolog (Brh2) serve as paradigms to understand the molecular actions of BRCA2 [53,54,55]. RecFOR and Brh2 recognize the ss/dsDNA junctions and load RecA and RAD51, respectively, onto the junctions to start one-directional filament growth from 5′ to 3′ [54,55,56]. Yeast Rad52 forms a multimeric ring-like structure and wraps ssDNA to destabilize RPA -ssDNA interaction for RAD51 loading [57,58,59]. The tower domain identified in the crystal structure of the BRCA2 CTD indicates a potential binding activity towards dsDNA [12], suggesting that BRCA2 could initiate a unidirectional RAD51 filament growth from an ss/dsDNA junction [12,53,55]. Unexpectedly, full-length BRCA2 displays no preference towards ss/dsDNA junctions and binds with almost equal affinity to 3′-tailed, 5′-tailed, and ssDNA, although tailed DNAs are the preferable substrates in RAD51-mediated strand exchange [18]. It is unknown whether some protein partners might endow structure-selective DNA-binding to BRCA2 towards ss/dsDNA junctions or other more complex DNA intermediates, potentially through altered DNA engagement of different DNA-binding domains (DBD). Overall, it is likely that the DNA-binding activities of BRCA2 and the potential for modulation by its protein partners might be uniquely fitted to its versatile activities in HR and fork stabilization.
Human RAD52 shows no mediator activity in standard reconstituted biochemical reactions [18], except at a sub-stoichiometric RAD51 concentration [73]. Deletion or depletion of RAD52 in mammalian cells causes no major HR defect, and it is generally accepted that BRCA2 is the major mediator in human cells [53,74]. However, unlike yeast Rad52, BRCA2 has no direct interaction with RPA but still stimulates RAD51 filament formation and DNA strand exchange activity in the presence of RPA inhibition [18,19]. More studies of BRCA2 are needed to determine how BRCA2 enables RAD51 filament formation without direct protein interaction with RPA .
Biochemical studies with individual BRC repeats demonstrate that BRC1-4 binds free RAD51 with high affinity, stimulates the formation of RAD51-ssDNA complexes, and prevents RAD51 binding onto dsDNA, while BRC5-8 stabilize RAD51-ssDNA nucleoprotein filaments [37,38]. An FxxA motif, identified from the high-resolution crystal structure of the BRC4-RAD51 complex, binds RAD51 by molecular mimicry of the oligomerization interface between individual RAD51 monomers [26]. The TR2 region was mapped to the extreme C-terminus as a secondary RAD51 binding site ( Figure 2 A). TR2 binds RAD51 filaments and regulates the function of BRCA2 in fork stabilization rather than HR through phosphorylation of the S3291 residue ( Figure 2 A) [27,28,29,79]. It remains unknown whether individual BRC repeats and the TR2 region cooperate in RAD51 recruitment, considering that the binding affinity of full-length BRCA2 to RAD51 is about 100-fold higher than that of an isolated BRC repeat [18]. Surprisingly, BRC5-8-DBD, a protein fragment containing BRC5-8 and the entire CTD (residues 1596–3418), shows enhanced stimulation in RAD51-catalyzed DNA strand exchange and almost full complementation of HR repair deficiency and RAD51 IR-induced foci formation in human BRCA2 knockout cell lines, compared to the same DBD constructs fused with a single BRC4, BRC repeats 1–4, or BRC repeats 1–8 [50]. It remains unclear whether BRC repeats could potentially antagonize each other, and further studies are needed to address these issues.
3. Role of BRCA2 in Replication Fork Restart
Stalled replication forks need to be restarted or converge with another fork to complete genome duplication before cells enter mitosis to prevent catastrophic DNA breakages and eventually cell death. Endogenous DNA lesions can trigger fork blockage or helicase-polymerase uncoupling and thus fork stalling, accompanied by the accumulation of ssDNA gaps [100,101]. Such DNA lesions include base damages, intra- and inter-strand crosslinks (ICLs), and protein-DNA complexes. Fork stalling lesions can also be induced by exogenous sources, such as UV radiation, aphidicolin, and MMC, or replication inhibitors such as hydroxyurea [100,101]. Cells utilize several different mechanisms to restart stalled forks, including fork reversal, fork breakage, translesion synthesis (TLS), and template switching [100,102]. This section will discuss the HR-dependent and HR-independent functions of BRCA2 in supporting replication fork restart, with a focus on fork reversal.
The function of BRCA2 in replication fork support had been linked to its stimulation of RAD51-mediated DNA strand invasion, a key reaction in the HR pathway to repair toxic intermediates produced when replication forks are stalled or broken, such as replication gaps and one-sided DSBs [100]. Surprisingly, recent data demonstrated a novel and HR-independent role of BRCA2 in stabilizing replication forks and protecting them against nuclease degradation [79]. This section will discuss recent progress focusing on the fork protection role of BRCA2 ( Figure 3 ).
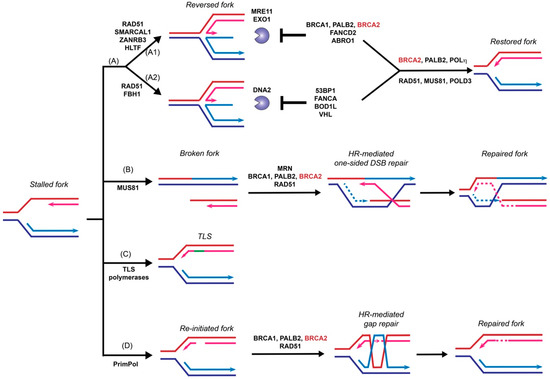
Fork reversal refers to the active remodeling of stalled replication forks after leading strand blockage, during which a so-called chicken foot structure can be formed by annealing the two newly synthesized sister strands leading to a DSB end ( Figure 3 ). This unique fork architecture was visualized by electron microscopy, occurring at an astounding frequency of 15–30% with all tested genotoxic treatments [101]. The high frequency of fork reversal substantiates its status as a likely physiological response to replication stress. There are two distinct pathways to remodel the forks actively and both require RAD51 but involve different motor proteins [103]. One route involves SMARCAL1 [104,105,106], ZRANB3 [104,107], and HLTF [108,109], all ATPase-dependent DNA translocases of the SWI2/SNF2 family, while the other involves the F-box DNA helicase FBH1, a UvrD homolog [110]. Reversed forks are targeted by multiple nucleases, including MRE11, EXO1, and DNA2 [79,111,112,113,114,115], and require fork protection to prevent nucleolytic degradation. As shown in Figure 3 , reversed forks from these two remodeling pathways are attacked by different nucleases and recruit distinct replication fork protection (RFP) factors [103]. BRCA2 is essential to protect the fork against MRE11-dependent degradation, together with FANCD2 and ABRO1 [103] ( Figure 3 A1). In BRCA2 -deficient cells, the replication fork undergoes massive degradation by MRE11 after HU treatment, as evidenced by DNA fiber assays [79,112,115], electron microscopic imaging [111], and 2D-gel electrophoresis [116]. Fork degradation could be suppressed by either BRCA2 back-complementation or MRE11 inhibition [79]. Depletion of SMARCAL1, ZRANB3, and HLTF, but not FBH1, restores fork stability in BRCA2 -deficient cells, suggesting that they participate in two different subpathways ( Figure 3 A) [103,112,117]. The recruitment of BRCA2 to stalled replication forks is dependent on BRCA1 and PALB2, as depletion of either one leads to fork destabilization [117,118,119]. Additionally, BRCA2 protects the fork from degradation by EXO1, but not DNA2, since knockdown of EXO1, but not DNA2, restores the fork stability in BRCA2 -deficient cells [111]. Instead, DNA2-mediated degradation is suppressed by 53BP1, FANCA, BOD1L, and VHL in the second pathway [103] ( Figure 3 A2). It is unclear what differentiates the two chicken foot pathways and whether there are alternate chicken foot structures.
Template switching relies on HR to bypass DNA lesions utilizing the newly synthesized daughter strand on the sister chromatid as a template for replication fork restart to repair postreplicative gaps [124] ( Figure 3 ). Moreover, extended treatment with hydroxyurea leads to prolonged blocking and breaking of replication forks, which requires the RAD51-mediated HR pathway to restart from one-sided DSBs [123]. MUS81 is responsible for generating these one-sided DSBs, and BRCA2 is expected to function as a mediator protein to facilitate RAD51 filament formation to catalyze homology search and DNA strand invasion ( Figure 3 B) [123,125,126]. An alternative pathway of stalled fork processing is fork repriming, which mainly relies on TLS polymerases to replicate through a damaged template ( Figure 3 C) or by skipping the lesion to reinitiate replisome downstream of the lesion by PrimPol in humans ( Figure 3 D) [100,102].
4. Pathogenic BRCA2 Mutations and BRCA2 Pseudo-Revertants
Loss-of-function mutations in the BRCA1 /2 genes are associated with increased genome instability and predisposition to different but not all forms of cancers. Defects in BRCA2 binding partners and other HR genes result in tumors with similar HR deficiency, termed as “BRCAness”. These tumors initially respond well to DNA damage-based therapies such as ionizing radiation, cisplatin, and PARPi. PARPi exploits the synthetic lethality with BRCA-deficiency (or BRCAness) leading to selective killing of tumor cells [226,227]. This section discusses pathogenic BRCA2 mutants and BRCA2 pseudo-revertants as genetic tools to understand the essential domains of BRCA2 in maintaining genome stability, which ultimately will facilitate future clinical diagnosis and patient treatment.
To date, over 20,000 variants of BRCA2 have been documented in the BRCA Exchange [228], and over 13,000 variants have been reported in the ClinVar database [47]. Almost 3000 variants of BRCA2 in the BRCA Exchange and about 1/3 of the variants in the ClinVar are considered to be pathogenic. Most pathogenic BRCA2 mutations are small insertions or deletions that result in a frameshift, generating a premature stop codon and producing a truncated protein similar to a nonsense mutation. The majority (86%) of the pathogenic variants are frameshift and nonsense mutation, while the rest are splicing and missense variants. Analysis of the mutation distribution among breast and ovarian cancer families identified several regions along the BRCA2 gene with high risks. The ovarian cancer cluster regions (OCCR) largely overlap with the BRC repeats and the helical domain of BRCA2 , whereas the breast cancer cluster regions (BCCR) cover both PALB2/EMSY-binding domains and DNA-binding domains [45,46]. Most of the pathogenic variants are located within these clusters, with the exception that some variants which are located in the C-terminal RAD51-binding domain ( Figure 2 ). The mechanistic basis of why different regions of BRCA2 are hotspots for distinct cancers remains unclear.
PARPi and platinum-based chemotherapy have been used to effectively treat BRCA2 mutant tumors. However, accumulating evidence indicates that tumors develop resistant mechanisms during treatment, and a clinically relevant mechanism is the acquisition of secondary mutations of BRCA2 to regain some degree of BRCA2 activity. These BRCA2 pseudo-revertants can arise from base substitutions and insertions or deletions with various sizes, resulting in a restoration of the BRCA2 open reading frame or removal of the initial deleterious mutations [229,230]. The pancreatic cancer cell line CAPAN-1 features the most prevalent BRCA2 mutation 6174delT lacking the C-terminal 1360 amino acids, including the last BRC repeat, the entire DBD, and the two NLSs [231]. Two of the PARP-inhibitor-resistant (PIR) clones identified from the CAPAN1 line, PIR1 and PIR2, have only recovered the extreme C-terminus containing the TR2 and NLS domains, compared to the original BRCA2 truncation mutant CAPAN1 6174delT [229]. Lacking the entire DBD and BRC6-8, PIR1 and PIR2 are still partly functional, as evidenced by restoration of some damage-induced RAD51 foci formation, reduced chromosomal aberrations, resistance to DNA damage agents, and loss of sensitivity towards PARPi [229]. It is likely that the PIR1 and PIR2 gained partial functions due to their intact protein interaction with PALB2, since the abolishment of PALB2 interaction completely inactivates a BRCA2 construct lacking the entire DBD in HR repair based on a DR-GFP assay [49]. It is unclear if additional genetic changes drive the phenotypes in these CAPAN1 pseudorevertants. The generation of pseudo-reversion mutations in BRCA2 -deficient tumors reflects tumor evolution under selection. There are at least three contributing factors: increased mutation rate due to exposure to genotoxic agents, the lack of error-free DNA repair, and a selective advantage for BRCA1 /2-restored cells during PARPi or platinum treatment [232]. Overall, the restored DNA repair function makes it challenging to target BRCA2 revertant tumors to overcome therapeutic resistance.
This entry is adapted from the peer-reviewed paper 10.3390/genes12081229