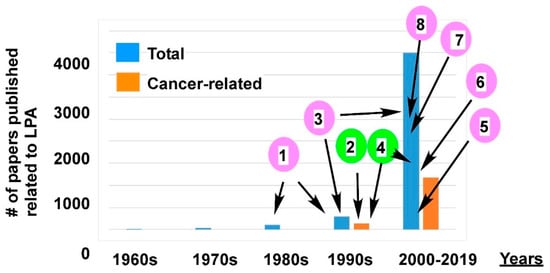
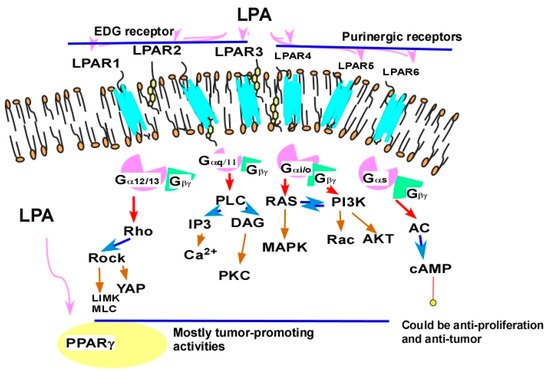
2.2.4.1. Cross-Talk between LPA Signaling and Other Cell Signaling Receptors
LPA elicits multiple and complex signaling pathways, which were extensively reviewed in recent years [
21,
31,
48,
61,
95,
206,
207,
208,
209]. LPA signaling pathways intertwine with almost all other major cell signaling pathways. We postulate that this network, instead of an individual LPA signaling pathway, represents a more effective target. Hence, this review focuses on LPA cross-talk with other signaling molecules. These molecules are often more “targetable” with FDA-approved inhibitors in clinical trials.
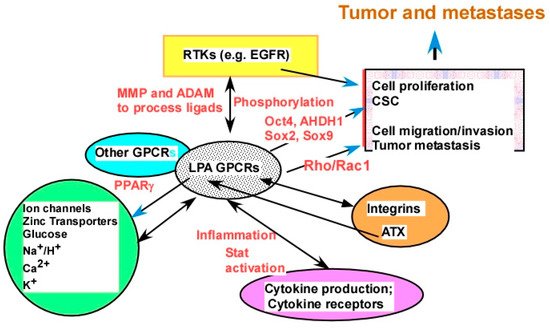
The cross-talk between LPA and other signaling molecules was extensively demonstrated, covering virtually every type of cell plasma membrane receptors, including ligand-gated ion channels, receptor tyrosine kinases (RTKs), receptors with other enzymatic activities (serine or serine/threonine kinases and guanylyl cyclase enzymatic activities), other GPCRs, integrins, cytokine receptors, and T- and B-cell receptors, as well as intracellular receptors, such as PPARγ. Listed below are examples from these categories (Figure 4).
Figure 4. LPA cross-talk as potential targets. LPA interacts with major types of plasma membrane receptors, including ion channels, metal ion transporters, other transporters, receptor tyrosine kinases (RTKs), other GPCRs, integrins, and cytokine receptors. Examples from each category of receptors are discussed in the
Section 2.2.4. Certain potential mechanisms of cross-talk are presented by words in red, including ligand production and/or processing, receptor phosphorylation, and production of downstream molecules mediating the cross-talk.
LPA stimulates and regulates several ion channels, including the Ca
2+ and Ca
2+-activated potassium channels, and the Na
+/H
+ exchanger 3 (NHE3) via the LPAR
5 receptor, which also involves the epidermal growth factor receptor (EGFR) [
210,
211]. LPA also regulates glucose transporters in skeletal muscle and adipose tissue [
212]. We recently showed that LPA upregulates ZIP4 (a zinc transporter) expression mainly via PPAR
γ [
133] (
Figure 4).
The cross-talk between receptor tyrosine kinase (RTK)–GPCR signal complexes is a focal point for the study of integration of cell signaling, which plays an important role in signal transduction [
213]. The cross-talk between LPA and EGFR is the best studied [
214,
215,
216,
217,
218,
219]. LPA also regulates and/or transactivates platelet derived growth factor receptor (PDGFR) [
220,
221,
222,
223,
224], tropomyosin receptor kinase A (TrkA), the receptors for nerve growth factor (NGF) [
225], Toll-like receptors [
226], and c-Met, the receptor for hepatocyte growth factor [
227].
LPA inhibits the natriuretic peptide-induced generation of cGMP via a non-receptor tyrosine kinase Csk [
228,
229,
230]. The best example of LPA’s effect on non-membrane receptors is its functions with regard to Src family kinases [
231,
232,
233,
234]. In addition, LPA regulates cytokines, such as IL-6, and its downstream signal transducers and activators of transcription (Stat) signaling molecules [
235].
LPA interacts with other GPCR receptors. Free fatty-acid receptors (FFARs; FFA1 and FFA4) have a potential negative cross-talk between LPA receptors and EGF receptors [
217,
236]. LPA stimulates endothelin (a GPCR ligand) expression and production in vascular smooth muscle cells [
237]. In addition, a cross-talk between the LPAR–G
13/p115RhoGEF/RhoA pathway and the β2-adrenergic receptor/G
s/adenylyl cyclase pathway was reported [
238]. LPA also cross-talks with α1 adrenoceptors [
239]. At physiological concentrations, LPA is capable of modulating opioid receptor binding [
240].
There are close interactions between two oncogenic lysolipids, LPA and S1P, in their overlapping signaling pathways and/or directly in their receptors [
241]. These two lipids can also cross-talk via ATX [
242,
243]. Transforming growth factor beta (TGFβ) may play a role in the LPA–S1P cross-talk [
244]. LPA upregulated expression of the cyclin-dependent kinase inhibitor p21(Waf1) in a TGFβ-dependent manner [
245]. Cross-talk between TNF-α and LPA results in the amplification of COX-2 protein expression via a conserved protein kinase D (PKD)-dependent signaling pathway [
246]. Hisano et al. used a genome-wide CRISPR/dCas9-based GPCR signaling screen to identify that LPAR
1 is an inducer of S1PR
1/β-arrestin coupling. This interaction promotes the porous junctional architecture of sinus-lining lymphatic endothelial cells and enables efficient lymphocyte trafficking [
247]. The functional link between LPA and integrins was established. Active integrin β1 is required for migration of fibroblastic cells [
248]. Laminin, but not other extracellular matrix proteins, induces LPA production in ovarian cancer cells via a β-integrin [
164]. LPA induces αvβ6-integrin-mediated TGFβ activation via the LPAR
2 and the small G
q [
249]. LPA upregulates integrins [
250,
251], and integrin signaling regulates the nuclear localization and function of the LPAR
1 in mammalian cells [
252]. Moreover, LPA-induced RhoA activation integrates the functions of integrins [
251,
253] and integrin α6β4 promotes expression of ATX in breast cancer cells [
254]. Most noticeably, ATX directly binds to several integrins [
91,
255], producing LPA close to the cell membrane [
256] (
Figure 4).
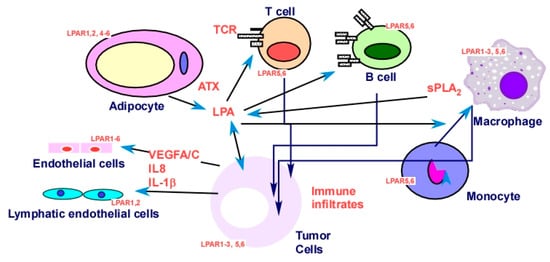
LPAR
5 functions as an inhibitory receptor able to negatively regulate T-cell receptor (TCR) signaling [
204]. LPAR
5 also inhibits B-cell receptor (BCR) signal transduction via a G
α12/13/Arhgef1 pathway [
257]. On the other hand, LPA augments IL-13 secretion from T cells via induction of submaximal T-cell activation [
258].
The cross-talk can be mono- or bidirectional and can be either positive or negative cross-talk, dependent on the type of interaction, the cell types, and the biological effects involved [
259]. For example, while LPA transactivates nerve growth factor signaling via the TrkA receptor, the latter also uses a G-protein-mediated mechanism to regulate the p42/p44 MAPK pathway [
260]. The bidirectional regulation between LPA and integrins is mentioned above (
Figure 4).
It is important to note that LPA is involved in several stem cell/cancer stem cell (CSC) signaling pathways. The ATX–LPA signaling pathway is recognized as a critical new player in CSC [
48]. LPA is involved in classical stemness pathways, such as the Wnt, Notch, and Hippo pathways [
189,
261,
262,
263,
264,
265].
2.2.4.2. The Molecular Mechanisms of LPA Cross-Talks
LPA cross-talks with other signaling molecules at many different levels with divergent mechanisms. Firstly, interactions are through direct binding/interactions. Homo- and heterodimerization of LPA/S1P receptors, ovarian cancer G protein coupled receptor-1 (OGR1) and GPR4, was shown using LacZ complementation assays [
266]. LPA receptors form homo- and heterodimers within the LPA receptor subgroup and heterodimers with other receptors, such as S1PR
1–3 and GPR4. Interestingly, it was shown that LPA remarkably enhances, through the LPAR
1/G
i protein, the OGR1-mediated vascular actions to acidic pH [
267]. These results suggest that targeting dimerization may be an effective way to block the signaling mediated by the receptors. Although GPCR dimerization was known for many years, this is an under-investigated area and warrants further investigation [
266] (
Figure 4).
Secondly, transactivation is mediated via enzymatic activities regulating phosphorylation and/or ligand processing. LPA induces EGF receptor transactivation through metalloproteinase (MMP) and a disintegrin and metalloproteinase (ADAM)-catalyzed membrane shedding of heparin-binding EGF and autocrine/paracrine activation of EGF [
231,
268,
269], and EGF can also modulate LPAR
1 function and the phosphorylation state [
268] (
Figure 4).
Thirdly, an LPA-regulated transcriptome is involved. LPA regulates many cytokines, including IL-6, IL-8, growth-regulated oncogene (GRO)-α [
19,
20,
270,
271], and cytokine leukemia inhibitory factor (LIF) [
241]. IL-6 mediates the LPA cross-talk between stromal and epithelial prostate cancer cells [
272]. LPA-induced macrophage migration inhibitory factor (MIF) promotes both tumor cell growth and angiogenesis via both the Ras/MAPK and Ras–Akt/PI3K signaling pathways [
273]. IL-6 exerts its biological activities through two molecules: IL-6R (IL-6 receptor) and gp130 [
274]. Moreover, gp130-mediated Janus kinase (JAK)/signal transducer and activation of transcription 3 (STAT3) is required for ATX expression in adipocytes [
177] (
Figure 4). LPA stimulates the expression of CSC-associated genes, including
OCT4,
SOX2,
SOX9,
ALDH1, and drug transporters [
133,
275,
276], with most of these gene products being functionally involved in CSC.
Fourthly, downstream signaling pathway interactions play important roles. The signaling pathways involved in LPA cross-talk include, but are not limited to the PI3K/Ras [
277], the mitogen-activated protein kinase (MAP kinase) [
277,
278], the focal adhesion [
119], the Wnt, integrin, the Rho/Rock, and the YAP pathways [
279,
280], reactive oxygen species (ROS), the DNA repair pathway, and the glycolytic pathway [
27], as well as the Rho–cAMP interaction [
281] (
Figure 4).
Finally, other signaling molecules may regulate metabolic enzymes for LPA and other lipid molecules. Neurotransmitters, cytokines, and growth factors regulate the activity of a key set of lipid-metabolizing enzymes, such as phospholipases, to affect LPA and other lipid signaling molecules [
282]. In addition, an acylglycerol kinase that produces LPA modulates cross-talk with EGFR in prostate cancer cells [
283].
The targeting of one or more of these cross-talks and/or the major LPA downstream signaling pathways may be critical and/or more efficient than targeting LPA or LPAR directly. For example, the FDA recently approved the first PI3K inhibitor for breast cancer treatment. The challenges are identifying one or more driver targets at the level of individual cancer type and individual patient.