Noncommunicable diseases, such as Alzheimer’s disease, breast and prostate cancer, are becoming increasingly prevalent in Western countries. To better elucidate the onset and evolution of these pathologies and ultimately design new preventive and therapeutic strategies, research activities focused on these biomedical areas have been supported by the European Union in the last two decades. While research has globally contributed increasing our understanding of the pathological mechanisms underlying these diseases, the failure rate in drug development still remains very high. Nowadays, it is important to monitor contribution to innovation and impact of funded research by means of defined indicators.
- biomedical research
- Alzheimer’s disease
- breast cancer
- prostate cancer
- funding
- indicators
- translational failure
- animal models
- cross-disciplinarity.
1. Definition
Dementia and cancer are becoming increasingly prevalent in Western countries. In the last two decades, research focused on Alzheimer’s disease (AD) and cancer, in particular, breast cancer (BC) and prostate cancer (PC), has been substantially funded both in Europe and worldwide. While scientific research outcomes have contributed to increase our understanding of the disease etiopathology, still the prevalence of these chronic degenerative conditions remains very high across the globe. By definition, no model is perfect. In particular, animal models of AD, BC, and PC have been and still are traditionally used in basic/fundamental, translational, and preclinical research to study human disease mechanisms, identify new therapeutic targets, and develop new drugs. However, animals do not adequately model some essential features of human disease; therefore, they are often unable to pave the way to the development of drugs effective in human patients. The rise of new technological tools and models in life science, and the increasing need for multidisciplinary approaches have encouraged many interdisciplinary research initiatives. With considerable funds being invested in biomedical research, it is becoming pivotal to define and apply indicators to monitor the contribution to innovation and impact of funded research.
2. Introduction
| Ranking | Cancer Type | New Cases Diagnosed in 2018 (both Sexes) | % of All Cancers (Excluding Non-Melanoma Skin Cancer) |
|---|---|---|---|
| 1 | Lung | 2,093,876 | 12.3 |
| 2 | Breast | 2,088,849 | 12.3 |
| 3 | Colorectal | 1,800,977 | 10.6 |
| 4 | Prostate | 1,276,106 | 7.5 |
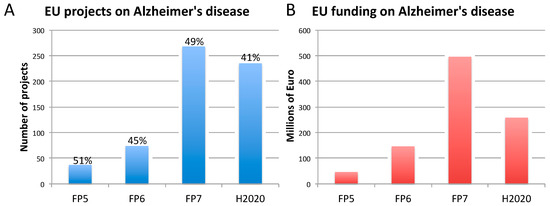
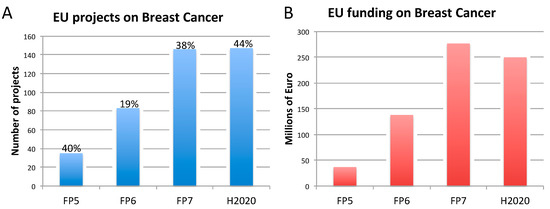
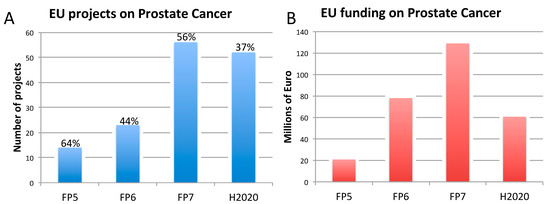
3. Three Biomedical Research Areas Characterized by a High Rate of Translational Failure: Alzheimer’s Disease, Breast Cancer, and Prostate Cancer
3.1. Alzheimer’s Disease
3.2. Breast Cancer
3.3. Prostate Cancer
References
- EC. Noncommunicable Diseases—NCDs. Available online: https://ec.europa.eu/knowledge4policy/foresight/topic/shifting-health-challenges/non-communicable-diseases-ncds_en (accessed on 23 April 2020).
- OECD. Health at a Glance: Europe 2018; OECD: Paris, France, 2018. [Google Scholar]
- Alzheimer’s Association. Alzheimer’s disease facts and figures. Alzheimer Dement 2019, 15, 321–387. [Google Scholar] [CrossRef]
- WHO. The Top 10 Causes of Death. Available online: https://http://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 23 April 2020).
- Deaths Registered in England and Wales (Series DR): 2017. Available online: https://http://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/bulletins/deathsregisteredinenglandandwalesseriesdr/2017 (accessed on 29 June 2020).
- Is Europe Ready for Alzheimer’s? Available online: http://www.aal-europe.eu/is-europe-ready-for-alzheimers/ (accessed on 29 June 2020).
- Worldwide Cancer Data. Available online: https://http://www.wcrf.org/dietandcancer/cancer-trends/worldwide-cancer-data (accessed on 23 April 2020).
- EC. Cancer Statistics—Specific Cancers. Available online: https://ec.europa.eu/eurostat/statistics-explained/pdfscache/39738.pdf (accessed on 29 June 2020).
- Simmons, D. The use of animal models in studying genetic disease: Transgenesis and induced mutation. Nat. Educ. 2008, 1, 70. [Google Scholar]
- Labant, M. Animal Models Evolve to Satisfy Emerging Needs. Available online: https://http://www.genengnews.com/insights/animal-models-evolve-to-satisfy-emerging-needs/ (accessed on 29 June 2020).
- Pound, P.; Ritskes-Hoitinga, M. Is it possible to overcome issues of external validity in preclinical animal research? Why most animal models are bound to fail. J. Transl. Med. 2018, 16, 304. [Google Scholar] [CrossRef]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar]
- Gould, S.E.; Junttila, M.R.; De Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K.; Williams, M. Preclinical Models of Alzheimer’s Disease: Relevance and Translational Validity. Curr. Protoc. Pharmacol. 2019, 84, e57. [Google Scholar] [CrossRef]
- Cavanaugh, S.E.; Pippin, J.J.; Barnard, N.D. Animal models of Alzheimer disease: Historical pitfalls and a path forward. Altex 2014, 31, 279–302. [Google Scholar] [CrossRef]
- Manning, H.C.; Buck, J.R.; Cook, R.S. Mouse Models of Breast Cancer: Platforms for Discovering Precision Imaging Diagnostics and Future Cancer Medicine. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2016, 57 (Suppl. 1), 60S–68S. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Sflomos, G.; Brisken, C. The challenges of modeling hormone receptor-positive breast cancer in mice. Endocr. -Relat. Cancer 2018, 25, R319–R330. [Google Scholar] [CrossRef]
- Rea, D.; Del Vecchio, V.; Palma, G.; Barbieri, A.; Falco, M.; Luciano, A.; De Biase, D.; Perdona, S.; Facchini, G.; Arra, C. Mouse Models in Prostate Cancer Translational Research: From Xenograft to PDX. Biomed. Res. Int. 2016, 2016, 9750795. [Google Scholar] [CrossRef] [PubMed]
- Hensley, P.J.; Kyprianou, N. Modeling prostate cancer in mice: Limitations and opportunities. J. Androl. 2012, 33, 133–144. [Google Scholar] [CrossRef] [PubMed]
- BIO, Biomedtracker, Amplion. Development Success Rates 2006–2015. Available online: https://www.bio.org/sites/default/files/legacy/bioorg/docs/Clinical%20Development%20Success%20Rates%202006-2015%20-%20BIO,%20Biomedtracker,%20Amplion%202016.pdf (accessed on 23 April 2020).
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef]
- Cummings, J.; Reiber, C.; Kumar, P. The price of progress: Funding and financing Alzheimer’s disease drug development. Alzheimer’s Dement. 2018, 4, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Miccoli, B.; Braeken, D.; Li, Y.E. Brain-on-a-chip Devices for Drug Screening and Disease Modeling Applications. Curr. Pharm. Des. 2018, 24, 5419–5436. [Google Scholar] [CrossRef]
- Eglen, R.M.; Reisine, T. Human iPS Cell-Derived Patient Tissues and 3D Cell Culture Part 2: Spheroids, Organoids, and Disease Modeling. Slas Technol. 2019, 24, 18–27. [Google Scholar] [CrossRef]
- Essayan-Perez, S.; Zhou, B.; Nabet, A.M.; Wernig, M.; Huang, Y.A. Modeling Alzheimer’s disease with human iPS cells: Advancements, lessons, and applications. Neurobiol. Dis. 2019, 130, 104503. [Google Scholar] [CrossRef]
- Shirotani, K.; Matsuo, K.; Ohtsuki, S.; Masuda, T.; Asai, M.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Kondo, T.; Inoue, H.; et al. A simplified and sensitive method to identify Alzheimer’s disease biomarker candidates using patient-derived induced pluripotent stem cells (iPSCs). J. Biochem. 2017, 162, 391–394. [Google Scholar] [CrossRef]
- Zanoni, M.; Pignatta, S.; Arienti, C.; Bonafe, M.; Tesei, A. Anticancer drug discovery using multicellular tumor spheroid models. Expert Opin. Drug Discov. 2019, 14, 289–301. [Google Scholar] [CrossRef]
- Lee, I.C. Cancer-on-a-chip for Drug Screening. Curr. Pharm. Des. 2018, 24, 5407–5418. [Google Scholar] [CrossRef] [PubMed]
- Silverman, E.K.; Schmidt, H.; Anastasiadou, E.; Altucci, L.; Angelini, M.; Badimon, L.; Balligand, J.L.; Benincasa, G.; Capasso, G.; Conte, F.; et al. Molecular networks in Network Medicine: Development and applications. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, e1489. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Hong, X.; Li, S.; Zhang, Y.; Zhao, Q.; Du, W.; Wang, Y.; Ni, J. Urine-Based Biomarkers for Alzheimer’s Disease Identified Through Coupling Computational and Experimental Methods. J. Alzheimer’s Dis. JAD 2018, 65, 421–431. [Google Scholar] [CrossRef] [PubMed]
- EC. FET Open. Available online: https://ec.europa.eu/programmes/horizon2020/en/h2020-section/fet-open (accessed on 23 April 2020).
- EC. Funding & Tender Opportunities. Available online: https://ec.europa.eu/info/funding-tenders/opportunities/portal/screen/opportunities/topic-search;freeTextSearchKeyword=;typeCodes=1;statusCodes=31094501,31094502;programCode=H2020;programDivisionCode=31047826;focusAreaCode=null;crossCuttingPriorityCode=null;callCode=Default;sortQuery=openingDate;orderBy=asc;onlyTenders=false;topicListKey=topicSearchTablePageState (accessed on 23 April 2020).
- Multidisciplinary Research Projects on Personalised Medicine—Pre-/Clinical Research, Big Data and ICT, Implementation and User’s Perspective. Available online: http://www.erapermed.eu/3211-2/ (accessed on 23 April 2020).
- IMI. Innovative Medicines Initiative. Available online: https://http://www.imi.europa.eu/ (accessed on 23 April 2020).
- EC. 2019 Report on the Statistics on the Use of Animals for Scientific Purposes in the Member States of the European Union in 2015–2017. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?qid=1581689520921&uri=CELEX:52020DC0016 (accessed on 23 April 2020).
- WHO. Ageing and Health. Available online: https://http://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on 23 April 2020).
- Dementia Statistics. Available online: https://http://www.alz.co.uk/research/statistics (accessed on 23 April 2020).
- Wimo, A.; Jonsson, L.; Bond, J.; Prince, M.; Winblad, B.; Alzheimer Disease, I. The worldwide economic impact of dementia 2010. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 9, 1–11 e13. [Google Scholar] [CrossRef] [PubMed]
- Early Onset Famlial, A.D. Available online: https://http://www.alzforum.org/early-onset-familial-ad/overview/what-early-onset-familial-alzheimer-disease-efad (accessed on 23 April 2020).
- Hunsberger, H.C.; Pinky, P.D.; Smith, W.; Suppiramaniam, V.; Reed, M.N. The role of APOE4 in Alzheimer’s disease: Strategies for future therapeutic interventions. Health Psychol. Behav. Med. 2019, 3, NS20180203. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Quintessential risk factors: Their role in promoting cognitive dysfunction and Alzheimer’s disease. Neurochem. Res. 2012, 37, 2627–2658. [Google Scholar] [CrossRef]
- World Alzheimer Report 2018. Available online: https://http://www.alz.co.uk/research/world-report-2018 (accessed on 23 April 2020).
- PCRM. Retiring the Amyloid Cascade Hypothesis as a Cause of Alzheimer’s. Available online: https://http://www.pcrm.org/news/good-science-digest/retiring-amyloid-cascade-hypothesis-cause-alzheimers (accessed on 23 April 2020).
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef]
- Makin, S. The amyloid hypothesis on trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef]
- Beach, T.G.; Monsell, S.E.; Phillips, L.E.; Kukull, W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J. Neuropathol. Exp. Neurol. 2012, 71, 266–273. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Qian, J.; Monsell, S.E.; Blacker, D.; Gomez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Johnson, K.A.; Frosch, M.P.; Sperling, R.A.; et al. Mild to moderate Alzheimer dementia with insufficient neuropathological changes. Ann. Neurol. 2014, 75, 597–601. [Google Scholar] [CrossRef]
- Pippin, J.J.; Cavanaugh, S.E.; Pistollato, F. Animal Research for Alzheimer Disease: Failures of Science and Ethics. In Animal Experimentation: Working Towards a Paradigm Change; Herrmann, K., Jayne, J., Eds.; Brill: Leiden, The Netherlands, 2019. [Google Scholar] [CrossRef]
- FDA-Approved Treatments for Alzheimer’s. Available online: https://http://www.alz.org/media/documents/fda-approved-treatments-alzheimers-ts.pdf (accessed on 23 April 2020).
- Birks, J. Cholinesterase Inhibitors for Alzheimer’s Disease. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 6, CD001190. [Google Scholar] [CrossRef] [PubMed]
- Shan, G.; Banks, S.; Miller, J.B.; Ritter, A.; Bernick, C.; Lombardo, J.; Cummings, J.L. Statistical advances in clinical trials and clinical research. Alzheimer’s Dement. 2018, 4, 366–371. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.; Van Der Graaf, P.H.; Arrowsmith, J.; Feltner, D.E.; Drummond, K.S.; Wegner, C.D.; Street, S.D. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov. Today 2012, 17, 419–424. [Google Scholar] [CrossRef]
- De Witte, W.E.A.; Danhof, M.; van der Graaf, P.H.; de Lange, E.C.M. The implications of target saturation for the use of drug-target residence time. Nat. Rev. Drug Discov. 2018, 18, 82–84. [Google Scholar] [CrossRef]
- Kleiman, R.J.; Ehlers, M.D. Data gaps limit the translational potential of preclinical research. Sci. Transl. Med. 2016, 8, 320–321. [Google Scholar] [CrossRef]
- Karran, E.; Hardy, J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann. Neurol. 2014, 76, 185–205. [Google Scholar] [CrossRef]
- Gold, M. Phase II clinical trials of anti-amyloid beta antibodies: When is enough, enough? Alzheimer’s Dement. 2017, 3, 402–409. [Google Scholar] [CrossRef]
- Gray, J.A.; Fleet, D.; Winblad, B. The need for thorough phase II studies in medicines development for Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 67. [Google Scholar] [CrossRef]
- Pagliarulo, N.; Gardner, J. 7 Questions on Biogen’s Revival of a Failed Alzheimer’s Drug. Available online: https://http://www.biopharmadive.com/news/biogen-alzheimers-aducanumab-revival-7-questions/565609/ (accessed on 23 April 2020).
- WHO. Risk Reduction of Cognitive Decline and Dementia; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Barrett, J.E.; McGonigle, P. Rodent Models for Alzheimer’s Disease in Drug Discovery. In Drug Discovery Approaches for the Treatment of Neurodegenerative Disorders; Academic Press: San Diego, CA, USA, 2017; pp. 235–247. [Google Scholar] [CrossRef]
- Newman, M.; Kretzschmar, D.; Khan, I.; Chen, M.; Verdile, G.; Lardelli, M. Animal models of Alzheimer’s Disease. In Animal Models for the Study of Human Disease, 2nd ed.; Michael Conn, P., Ed.; Academic Press: San Diego, CA, USA, 2017; pp. 1031–1085. [Google Scholar]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Research Models. Alzheimer’s Disease. Available online: https://http://www.alzforum.org/research-models/alzheimers-disease (accessed on 23 April 2020).
- King, A. The search for better animal models of Alzheimer’s disease. Nature 2018, 559, S13–S15. [Google Scholar] [CrossRef] [PubMed]
- Pallàs, M. Senescence-Accelerated Mice P8: A Tool to Study Brain Aging and Alzheimer’s Disease in a Mouse Model. Int. Sch. Res. Not. Cell Biol. 2012, 2012, 1–12. [Google Scholar] [CrossRef]
- Cheng, X.R.; Zhou, W.X.; Zhang, Y.X. The behavioral, pathological and therapeutic features of the senescence-accelerated mouse prone 8 strain as an Alzheimer’s disease animal model. Ageing Res. Rev. 2014, 13, 13–37. [Google Scholar] [CrossRef]
- Lok, K.; Zhao, H.; Zhang, C.; He, N.; Shen, H.; Wang, Z.; Zhao, W.; Yin, M. Effects of accelerated senescence on learning and memory, locomotion and anxiety-like behavior in APP/PS1 mouse model of Alzheimer’s disease. J. Neurol. Sci. 2013, 335, 145–154. [Google Scholar] [CrossRef] [PubMed]
- App KO/APOE4/Trem2*R47H. Available online: https://http://www.jax.org/strain/031722 (accessed on 23 April 2020).
- Dong, J.; Zhou, M.; Wu, X.; Du, M.; Wang, X. Memantine combined with environmental enrichment improves spatial memory and alleviates Alzheimer’s disease-like pathology in senescence-accelerated prone-8 (SAMP8) mice. J. Biomed. Res. 2012, 26, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Liu, G.; Shi, S.; Li, Y.; Li, Z. Effects of manual acupuncture combined with donepezil in a mouse model of Alzheimer’s disease. Acupunct. Med. J. Br. Med Acupunct. Soc. 2019, 37, 64–71. [Google Scholar] [CrossRef]
- Folch, J.; Busquets, O.; Ettcheto, M.; Sanchez-Lopez, E.; Castro-Torres, R.D.; Verdaguer, E.; Garcia, M.L.; Olloquequi, J.; Casadesus, G.; Beas-Zarate, C.; et al. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J. Alzheimer’s Dis. JAD 2018, 62, 1223–1240. [Google Scholar] [CrossRef]
- Grossberg, G.T.; Manes, F.; Allegri, R.F.; Gutierrez-Robledo, L.M.; Gloger, S.; Xie, L.; Jia, X.D.; Pejovic, V.; Miller, M.L.; Perhach, J.L.; et al. The safety, tolerability, and efficacy of once-daily memantine (28 mg): A multinational, randomized, double-blind, placebo-controlled trial in patients with moderate-to-severe Alzheimer’s disease taking cholinesterase inhibitors. CNS Drugs 2013, 27, 469–478. [Google Scholar] [CrossRef]
- Howard, R.; McShane, R.; Lindesay, J.; Ritchie, C.; Baldwin, A.; Barber, R.; Burns, A.; Dening, T.; Findlay, D.; Holmes, C.; et al. Nursing home placement in the Donepezil and Memantine in Moderate to Severe Alzheimer’s Disease (DOMINO-AD) trial: Secondary and post-hoc analyses. Lancet. Neurol. 2015, 14, 1171–1181. [Google Scholar] [CrossRef]
- Porquet, D.; Andres-Benito, P.; Grinan-Ferre, C.; Camins, A.; Ferrer, I.; Canudas, A.M.; Del Valle, J.; Pallas, M. Amyloid and tau pathology of familial Alzheimer’s disease APP/PS1 mouse model in a senescence phenotype background (SAMP8). Age 2015, 37, 9747. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-beta in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef]
- Veening-Griffioen, D.H.; Ferreira, G.S.; van Meer, P.J.K.; Boon, W.P.C.; Gispen-de Wied, C.C.; Moors, E.H.M.; Schellekens, H. Are some animal models more equal than others? A case study on the translational value of animal models of efficacy for Alzheimer’s disease. Eur. J. Pharmacol. 2019, 859, 172524. [Google Scholar] [CrossRef] [PubMed]
- Rae, E.A.; Brown, R.E. The problem of genotype and sex differences in life expectancy in transgenic AD mice. Neurosci. Biobehav. Rev. 2015, 57, 238–251. [Google Scholar] [CrossRef]
- Flurkey, K.; Currer, J.M.; Harrison, D.E. Mouse Models in Aging Research. In The Mouse in Biomedical Research, 2nd ed.; Medicine, A., Ed.; Academic Press: Cambridge, MA, USA, 2007; Volume III, pp. 637–672. [Google Scholar]
- Yuan, R.; Peters, L.L.; Paigen, B. Mice as a mammalian model for research on the genetics of aging. Ilar J. 2011, 52, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer: Statistics. Available online: https://http://www.cancer.net/cancer-types/breast-cancer/statistics (accessed on 24 April 2020).
- cancer.org. Survival Rates for Breast Cancer. Available online: http://www.cancer.org/cancer/breastcancer/detailedguide/breast-cancer-survival-by-stage (accessed on 24 April 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- NIH. Drugs Approved for Breast Cancer. Available online: https://http://www.cancer.gov/about-cancer/treatment/drugs/breast (accessed on 23 April 2020).
- Leo, C.P.; Leo, C.; Szucs, T.D. Breast cancer drug approvals by the US FDA from 1949 to 2018. Nat. Rev. Drug Discov. 2020, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L.; Kirk, R. High drug attrition rates--where are we going wrong? Nat. Rev. Clin. Oncol. 2011, 8, 189–190. [Google Scholar] [CrossRef]
- Ju, J.; Zhu, A.J.; Yuan, P. Progress in targeted therapy for breast cancer. Chronic Dis. Transl. Med. 2018, 4, 164–175. [Google Scholar] [CrossRef]
- Fan, P.; Jordan, V.C. New insights into acquired endocrine resistance of breast cancer. Cancer Drug Resist. 2019, 2, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Guo, J.; Shen, B.Q.; Yadav, D.B.; Sliwkowski, M.X.; Crocker, L.M.; Lacap, J.A.; Phillips, G.D.L. Mechanisms of Acquired Resistance to Trastuzumab Emtansine in Breast Cancer Cells. Mol. Cancer Ther. 2018, 17, 1441–1453. [Google Scholar] [CrossRef]
- Luque-Cabal, M.; Garcia-Teijido, P.; Fernandez-Perez, Y.; Sanchez-Lorenzo, L.; Palacio-Vazquez, I. Mechanisms Behind the Resistance to Trastuzumab in HER2-Amplified Breast Cancer and Strategies to Overcome It. Clin. Med. Insights Oncol. 2016, 10, 21–30. [Google Scholar] [CrossRef]
- Moiseenko, F.; Volkov, N.; Bogdanov, A.; Dubina, M.; Moiseyenko, V. Resistance mechanisms to drug therapy in breast cancer and other solid tumors: An opinion. F1000Research 2017, 6, 288. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. New Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- Mateo, J.; Ong, M.; Tan, D.S.; Gonzalez, M.A.; de Bono, J.S. Appraising iniparib, the PARP inhibitor that never was—What must we learn? Nat. Rev. Clin. Oncol. 2013, 10, 688–696. [Google Scholar] [CrossRef]
- Whittle, J.R.; Lewis, M.T.; Lindeman, G.J.; Visvader, J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. BCR 2015, 17, 17. [Google Scholar] [CrossRef]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The Landscape of Targeted Therapies in TNBC. Cancers 2020, 12. [Google Scholar] [CrossRef]
- NIH. The Cancer Genome Atlas Program. Available online: https://http://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 24 April 2020).
- Holen, I.; Speirs, V.; Morrissey, B.; Blyth, K. In vivo models in breast cancer research: Progress, challenges and future directions. Dis. Models Mech. 2017, 10, 359–371. [Google Scholar] [CrossRef]
- Eccles, S.A.; Aboagye, E.O.; Ali, S.; Anderson, A.S.; Armes, J.; Berditchevski, F.; Blaydes, J.P.; Brennan, K.; Brown, N.J.; Bryant, H.E.; et al. Critical research gaps and translational priorities for the successful prevention and treatment of breast cancer. Breast Cancer Res. BCR 2013, 15, R92. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Gisselsson, D.; Lichtenzstejn, D.; Kachko, P.; Karlsson, J.; Manor, E.; Mai, S. Clonal evolution through genetic bottlenecks and telomere attrition: Potential threats to in vitro data reproducibility. GenesChromosomes Cancer 2019, 58, 452–461. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Fleming, J.M.; Miller, T.C.; Meyer, M.J.; Ginsburg, E.; Vonderhaar, B.K. Local regulation of human breast xenograft models. J. Cell. Physiol. 2010, 224, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Fung, A.S.; Lee, C.; Yu, M.; Tannock, I.F. The effect of chemotherapeutic agents on tumor vasculature in subcutaneous and orthotopic human tumor xenografts. BMC Cancer 2015, 15, 112. [Google Scholar] [CrossRef] [PubMed]
- Cell Line-Derived Xenograft—CDX Model Studies in Mice. Available online: https://http://www.criver.com/products-services/discovery-services/pharmacology-studies/oncology-immuno-oncology-studies/oncology-models/cell-line-derived-xenograft-cdx-mouse-models?region=3696 (accessed on 29 June 2020).
- Cell-Line-Derived Xenograft Models. Available online: https://http://www.creative-animodel.com/animal-model-development/cell-line-derived-xenograft-models.html (accessed on 29 June 2020).
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, A.; Wang, L. Bridging the divide: Preclinical research discrepancies between triple-negative breast cancer cell lines and patient tumors. Oncotarget 2017, 8, 113269–113281. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, S.; Kim, K.; Kim, S.H.; Chung, Y.J.; Lee, C. Studying cancer immunotherapy using patient-derived xenografts (PDXs) in humanized mice. Exp. Mol. Med. 2018, 50, 99. [Google Scholar] [CrossRef]
- Brehm, M.A.; Shultz, L.D.; Luban, J.; Greiner, D.L. Overcoming current limitations in humanized mouse research. J. Infect. Dis. 2013, 208 (Suppl. 2), S125–S130. [Google Scholar] [CrossRef]
- Garcia, S.; Freitas, A.A. Humanized mice: Current states and perspectives. Immunol. Lett. 2012, 146, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Laudanski, K.; Stentz, M.; DiMeglio, M.; Furey, W.; Steinberg, T.; Patel, A. Potential Pitfalls of the Humanized Mice in Modeling Sepsis. Int. J. Inflamm. 2018, 2018, 6563454. [Google Scholar] [CrossRef] [PubMed]
- Yeadon, J. Immunodeficient Mice for Cancer Studies: Which Host Strain Should I Use? Available online: https://http://www.jax.org/news-and-insights/jax-blog/2013/july/which-host-strain-should-i-use (accessed on 29 June 2020).
- Eyre, R.; Alferez, D.G.; Spence, K.; Kamal, M.; Shaw, F.L.; Simoes, B.M.; Santiago-Gomez, A.; Sarmiento-Castro, A.; Bramley, M.; Absar, M.; et al. Patient-derived Mammosphere and Xenograft Tumour Initiation Correlates with Progression to Metastasis. J. Mammary Gland Biol. Neoplasia 2016, 21, 99–109. [Google Scholar] [CrossRef] [PubMed]
- STOCK Trp53tm1Brd Brca1tm1Aash Tg(LGB-cre)74Acl/J. Available online: https://http://www.jax.org/strain/012620 (accessed on 29 June 2020).
- Menezes, M.E.; Das, S.K.; Emdad, L.; Windle, J.J.; Wang, X.Y.; Sarkar, D.; Fisher, P.B. Genetically engineered mice as experimental tools to dissect the critical events in breast cancer. Adv. Cancer Res. 2014, 121, 331–382. [Google Scholar] [CrossRef] [PubMed]
- Dabydeen, S.A.; Furth, P.A. Genetically engineered ERalpha-positive breast cancer mouse models. Endocr. -Relat. Cancer 2014, 21, R195–R208. [Google Scholar] [CrossRef] [PubMed]
- Greenow, K.R.; Smalley, M.J. Overview of Genetically Engineered Mouse Models of Breast Cancer Used in Translational Biology and Drug Development. Curr. Protoc. Pharmacol. 2015, 70, 14.36. 1–14.36. 14. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Khadka, P.; Jin, X.; Wong, B.; Franke, L.; Golub, T.R. The landscape of chromosomal aberrations in breast cancer mouse models reveals driver-specific routes to tumorigenesis. Nat. Commun. 2016, 7, 12160. [Google Scholar] [CrossRef]
- Fry, E.A.; Taneja, P.; Inoue, K. Oncogenic and tumor-suppressive mouse models for breast cancer engaging HER2/neu. Int. J. Cancer 2017, 140, 495–503. [Google Scholar] [CrossRef]
- Sledge, G.W., Jr. Curing Metastatic Breast Cancer. J. Oncol. Pract. 2016, 12, 6–10. [Google Scholar] [CrossRef]
- Estimates for Funding for Research for Metastatic Disease: LOW. Available online: http://mbcn.org/research-funding/ (accessed on 12 May 2020).
- EC. CORDIS. Available online: https://cordis.europa.eu/projects/en (accessed on 23 April 2020).
- Milosevic, M.; Jankovic, D.; Milenkovic, A.; Stojanov, D. Early diagnosis and detection of breast cancer. Technol. Health Care Off. J. Eur. Soc. Eng. Med. 2018, 26, 729–759. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, J.; Sun, Y. Circulating Tumor DNA as Biomarkers for Cancer Detection. Genom. Proteom. Bioinform. 2017, 15, 59–72. [Google Scholar] [CrossRef]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Taitt, H.E. Global Trends and Prostate Cancer: A Review of Incidence, Detection, and Mortality as Influenced by Race, Ethnicity, and Geographic Location. Am. J. Men’s Health 2018, 12, 1807–1823. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Abate-Shen, C.; Agus, D.B.; Attar, R.M.; Chung, L.W.; Greenberg, N.M.; Hahn, W.C.; Isaacs, J.T.; Navone, N.M.; Peehl, D.M.; et al. The current state of preclinical prostate cancer animal models. Prostate 2008, 68, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Ittmann, M.; Huang, J.; Radaelli, E.; Martin, P.; Signoretti, S.; Sullivan, R.; Simons, B.W.; Ward, J.M.; Robinson, B.D.; Chu, G.C.; et al. Animal models of human prostate cancer: The consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013, 73, 2718–2736. [Google Scholar] [CrossRef]
- Cho, H.; Herzka, T.; Zheng, W.; Qi, J.; Wilkinson, J.E.; Bradner, J.E.; Robinson, B.D.; Castillo-Martin, M.; Cordon-Cardo, C.; Trotman, L.C. RapidCaP, a novel GEM model for metastatic prostate cancer analysis and therapy, reveals myc as a driver of Pten-mutant metastasis. Cancer Discov. 2014, 4, 318–333. [Google Scholar] [CrossRef]
- Hsieh, C.L.; Xie, Z.; Liu, Z.Y.; Green, J.E.; Martin, W.D.; Datta, M.W.; Yeung, F.; Pan, D.; Chung, L.W. A luciferase transgenic mouse model: Visualization of prostate development and its androgen responsiveness in live animals. J. Mol. Endocrinol. 2005, 35, 293–304. [Google Scholar] [CrossRef]
- Aggarwal, S.; Ricklis, R.M.; Williams, S.A.; Denmeade, S.R. Comparative study of PSMA expression in the prostate of mouse, dog, monkey, and human. Prostate 2006, 66, 903–910. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; Pienta, K.J. Drug discovery in prostate cancer mouse models. Expert Opin. Drug Discov. 2015, 10, 1011–1024. [Google Scholar] [CrossRef]
- Wei, C.; Willis, R.A.; Tilton, B.R.; Looney, R.J.; Lord, E.M.; Barth, R.K.; Frelinger, J.G. Tissue-specific expression of the human prostate-specific antigen gene in transgenic mice: Implications for tolerance and immunotherapy. Proc. Natl. Acad. Sci. USA. 1997, 94, 6369–6374. [Google Scholar] [CrossRef] [PubMed]
- Bullock, L.P. Brief overview of selected aspects of testicular hormone action. Environ. Health Perspect. 1981, 38, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.S.; Dzinic, S.; Bonfil, A.I.; Saliganan, A.D.; Sheng, S.; Bonfil, R.D. The mouse prostate: A basic anatomical and histological guideline. Bosn. J. Basic Med Sci. 2016, 16, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Michiel Sedelaar, J.P.; Dalrymple, S.S.; Isaacs, J.T. Of mice and men—warning: Intact versus castrated adult male mice as xenograft hosts are equivalent to hypogonadal versus abiraterone treated aging human males, respectively. Prostate 2013, 73, 1316–1325. [Google Scholar] [CrossRef]
- Jeet, V.; Russell, P.J.; Khatri, A. Modeling prostate cancer: A perspective on transgenic mouse models. Cancer Metastasis Rev. 2010, 29, 123–142. [Google Scholar] [CrossRef]
- Kasper, S. Survey of genetically engineered mouse models for prostate cancer: Analyzing the molecular basis of prostate cancer development, progression, and metastasis. J. Cell. Biochem. 2005, 94, 279–297. [Google Scholar] [CrossRef]
- Kido, L.A.; de Almeida Lamas, C.; Marostica, M.R., Jr.; Cagnon, V.H.A. Transgenic Adenocarcinoma of the Mouse Prostate (TRAMP) model: A good alternative to study PCa progression and chemoprevention approaches. Life Sci. 2019, 217, 141–147. [Google Scholar] [CrossRef]
- Civenni, G.; Carbone, G.M.; Catapano, C.V. Overview of Genetically Engineered Mouse Models of Prostate Cancer and Their Applications in Drug Discovery. Curr. Protoc. Pharmacol. 2018, 81, e39. [Google Scholar] [CrossRef]
- Irshad, S.; Abate-Shen, C. Modeling prostate cancer in mice: Something old, something new, something premalignant, something metastatic. Cancer Metastasis Rev. 2013, 32, 109–122. [Google Scholar] [CrossRef]
- Lin, D.; Wyatt, A.W.; Xue, H.; Wang, Y.; Dong, X.; Haegert, A.; Wu, R.; Brahmbhatt, S.; Mo, F.; Jong, L.; et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014, 74, 1272–1283. [Google Scholar] [CrossRef]
- Hara, T.; Kouno, J.; Kaku, T.; Takeuchi, T.; Kusaka, M.; Tasaka, A.; Yamaoka, M. Effect of a novel 17,20-lyase inhibitor, orteronel (TAK-700), on androgen synthesis in male rats. J. Steroid Biochem. Mol. Biol. 2013, 134, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, M.; Hara, T.; Hitaka, T.; Kaku, T.; Takeuchi, T.; Takahashi, J.; Asahi, S.; Miki, H.; Tasaka, A.; Kusaka, M. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: Effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J. Steroid Biochem. Mol. Biol. 2012, 129, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Van Hook, K.; Huang, T.; Alumkal, J.J. Orteronel for the treatment of prostate cancer. Future Oncol. 2014, 10, 803–811. [Google Scholar] [CrossRef] [PubMed]
- NIH. Study to Investigate the Effects of Orteronel on the QT/QTc Interval in Patients with Metastatic Castration-Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01549951?view=results (accessed on 23 April 2020).
- NIH. Study Comparing Orteronel Plus Prednisone in Participants with Metastatic Castration-Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01193257 (accessed on 23 April 2020).
- Loddick, S.A.; Ross, S.J.; Thomason, A.G.; Robinson, D.M.; Walker, G.E.; Dunkley, T.P.; Brave, S.R.; Broadbent, N.; Stratton, N.C.; Trueman, D.; et al. AZD3514: A small molecule that modulates androgen receptor signaling and function in vitro and in vivo. Mol. Cancer Ther. 2013, 12, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Therapeutic Rationales, Progresses, Failures, and Future Directions for Advanced Prostate Cancer. Int. J. Biol. Sci. 2016, 12, 409–426. [Google Scholar] [CrossRef]
- Suzman, D.L.; Antonarakis, E.S. Castration-resistant prostate cancer: Latest evidence and therapeutic implications. Ther. Adv. Med Oncol. 2014, 6, 167–179. [Google Scholar] [CrossRef]
- NIH. Drugs Approved for Prostate Cancer. Available online: https://http://www.cancer.gov/about-cancer/treatment/drugs/prostate (accessed on 23 April 2020).
This entry is adapted from the peer-reviewed paper 10.3390/ani10071194