2.1. Current Therapeutic Approach
Hepatocellular carcinoma (HCC) is a primary tumor of the liver, often arising from cirrhosis, and is frequently associated with chronic hepatitis B infection, chronic hepatitis C infection, and alcoholism [
7,
8]. HCC is the most common primary liver tumor, making up 80–90% of all primary liver neoplasms. Approximately 900,000 new cases of liver cancer were reported in 2020 and liver cancer was the third leading cause of cancer-related deaths worldwide in the same year [
9]. In the United States, liver cancer has a 5-year survival rate of 18%, making it the second deadliest site of cancer [
6,
10].
Treatment of HCC is dictated by the Child–Pugh class and/or the Barcelona Clinic Liver Cancer staging system (BCLC). Early-stage HCC that is confined to the liver is primarily treated with surgical resection and/or ablation, which seek to completely remove the tumor [
11]. Transplantation is another therapeutic option that removes both the HCC as well as the underlying cirrhotic liver [
6,
12,
13]. Despite the curative intent of these treatment modalities, tumors frequently recur and the 5-year recurrence after surgical resection can be as high as 40–70%, and recurrence after transplantation can be as high as 20–40% [
6,
14]. In addition, since many patients with HCC present with advanced disease or complications related to cirrhosis, only 5–10% of HCC patients are candidates for surgical therapy [
15]. Transarterial therapies including transarterial chemoembolization (TACE) and selective internal radiation therapy (SIRT) can be considered for patients with unresectable, intermediate-staged BCLC B HCC [
6]. While these approaches can be helpful, their efficacy as a monotherapy has been limited.
Effective systemic therapies could potentially prolong the survival of patients with HCC. HCC, however, has been traditionally resistant to conventional cytotoxic chemotherapy, and only recently have small-molecule inhibitors been used. In 2008, sorafenib was tested in a phase III trial in patients with advanced HCC and was found to be associated with both an improvement in survival and delay in radiologic progression of 3 months versus placebo [
16,
17]. As a result, sorafenib was the first systemic therapy approved for the treatment of HCC. Other small-molecule inhibitors including lenvatinib, regorafenib, cabozantinib, and ramucirumab have been developed for HCC [
6,
18,
19,
20,
21]. Of the newly approved molecules, only sorafenib, lenvatinib, and regorafenib have been associated with an increased median overall survival to 6.5–13.6 months depending on the study [
15,
16,
17,
18,
19]. While this is an incremental improvement, the median survival for patients on these therapies remains rather dismal, highlighting the need for more efficacious systemic therapies. Given the success of immunotherapy in the treatment of some cancers, more recent efforts have been aimed at translating this success to patients with HCC.
2.2. The Challenging Tumor Microenvironment of HCC
The development and testing of immunotherapies among patients with HCC have taken significantly longer than in other cancers such as melanoma, mismatch repair-deficient colon cancer, or even lung cancer. While the reason for this is multifactorial, it likely is related to the reduced immunogenicity of HCC tumors. Compared with other types of tumors, such as melanoma, the tumor mutational burden of HCC tends to be low to moderate. With fewer somatic mutations within the tumor, there are a reduced number of tumor-specific neoantigens available to drive an adaptive immune response [
22]. Tumor mutational burden has been used as a biomarker to indicate the efficacy of T cells against a tumor, with a lower tumor mutational burden indicating a weaker immune response [
23]. In one study of 33 patients with HCC, the median TMB was 5.48 (range 1.68–16.07) and did not correlate with pathologic features of HCC [
24]. More so, the impact of the tumor mutational burden in HCC on the adaptive immune response, and its relation to tumor progression and patient outcome, is poorly understood [
3]. Taken together, these unique aspects to HCC have made it much more difficult to treat with immunotherapy and, importantly, have resulted in a drastic delay in the development and use of immunotherapy for patients with HCC.
Furthermore, the liver naturally creates an immunosuppressive microenvironment that facilitates tumor development. The liver provides an “immune-tolerant” environment due to its need to be accepting of new antigens encountered from food and microbial antigens delivered from the gastrointestinal tract [
25]. Tolerance of antigens is partially achieved through myeloid-derived suppressor cells that secrete chemokines, cytokines, and growth factors, which increase proliferation, protect from apoptosis, increase angiogenesis, and support growth [
26]. In addition, the activation and accumulation of regulatory T cells alongside upregulated programmed cell death protein 1 ligand (PD-L1) may contribute to the immunosuppressive environment. Lastly, these anti-inflammatory mediators can be increased in patients with cirrhosis [
27,
28,
29], further contributing to the immunosuppressive tumor microenvironment of the liver. Taken together, the reduced mutational burden of primary HCC and the immunosuppressive tumor microenvironment have created a hostile landscape for the development and testing of novel immunotherapies for the treatment of HCC.
2.3. Immune Checkpoint Inhibitors
The first immune checkpoint inhibitor (ICI) clinical trials for HCC began in 2008 [
30]. These early ICIs targeted cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4). It was discovered that CTLA-4 altered intracellular T cell signaling and blocked CD28 binding to CD80 and CD86, which is needed for optimal T cell activation [
31,
32,
33]. As a result, CTLA-4 decreases helper T cell activity and increases regulatory T cell activity [
34], which further inhibits the adaptive immune response to cancer. Blocking CTLA-4 with antibodies in mouse models allowed for improved antitumor activity [
35,
36]. Clinical trials conducted in 2009 demonstrated that blocking CTLA-4 with monoclonal antibodies improved survival in the treatment of patients with malignant melanoma [
37]. Eventually, the effects of CTLA-4 were studied in other cancers, including HCC. A phase II clinical trial of tremelimumab, an anti-CTLA-4 monoclonal antibody, in patients with hepatitis C-induced, advanced HCC achieved a partial response or stable disease in 76.4% of patients and had the added benefit of reducing hepatitis C viral load [
30]. Despite these findings, the survival of patients treated with tremilimumab was only 43% at one year. While these early efforts confirmed that immunotherapy could be used to treat patients with HCC, subsequent clinical trials with more novel checkpoint inhibitors would help to solidify the role of immunotherapy as a treatment for HCC. Soon after the discovery of CTLA-4, another immune checkpoint inhibitor was discovered. The binding of the T cell surface receptor, programmed death receptor-1 (PD-1), to its ligand located on tumor cells, called programmed death ligand 1 (PD-L1), was found to inhibit T cell-mediated cytotoxicity when bound to tumor cells [
34]. Early studies suggested that blockade of this interaction would improve T cell-induced immunity against cancer by disinhibiting the T cells and facilitating improved cytotoxicity [
38,
39,
40].
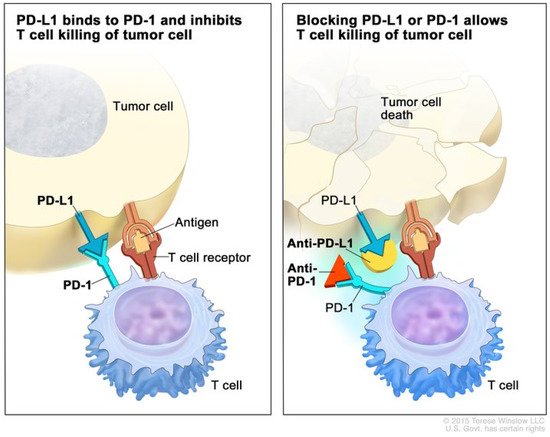
Figure 1 illustrates the mechanism of anti-PD-1 therapy. Clinical trials in patients with melanoma, colorectal cancer, prostate cancer, non-small-cell lung cancer, and renal cell carcinoma established the benefit of the PD-1/PD-L1 blockade in humans and have revolutionized the treatment of patients with these cancers [
41].
Figure 1. Mechanism of PD-1 and PD-L1 immune checkpoint inhibitors. (Left) The binding of PD-1 on the T cell to PD-L1 on the tumor cell inhibits effector T cell function, despite binding of the T cell receptor to an antigen. (Right) Blockade of PD-1 or PD-L1 with monoclonal antibodies prevents PD-1 binding with PD-L1 and promotes effector T cell function, leading to apoptosis of the tumor cell. Used with permission from Terrese Winslow LLC.
Studies of the immune landscape within the TME of HCC have found that exhausted CD8+ T cells (those that overexpress PD-1) were both preferentially enriched and potentially clonally expanded. As such, the PD-1/PDL-1 axis represents an important pathway to target in order to induce an immune response in patients with HCC [
42]. In 2016 and 2017, anti-PD1/PD-L1 monoclonal antibodies were studied in advanced, unresectable HCC. In two phase II trials, nivolumab and pembrolizumab prolonged survival and resulted in a 15–20% objective response rate (ORR), of which there was one (1%) complete and 17 (16%) partial responses in the KEYNOTE-224 [
43,
44]. These findings were confirmed in two additional phase III clinical trials. In the study by Finn et al., second-line therapy with pembrolizumab following treatment with sorafenib resulted in reduced progression in advanced HCC over two years versus placebo. The study also demonstrated prolonged survival; however, despite the results being significantly different from placebo, the prolonged survival failed to meet the statistical threshold set by the investigators [
3,
45]. Likewise, a phase III trial comparing nivolumab to sorafenib in patients with advanced HCC resulted in an improvement in both 1-year and 2-year survival. However, with a minimum follow-up of 22.8 months, overall survival was not significantly different between the two cohorts [
46]. In contrast, longer-term studies with a minimum follow-up of 33.6 months resulted in improved safety and survival among patients treated with nivolumab compared with sorafenib when used in the setting of advanced HCC [
47].
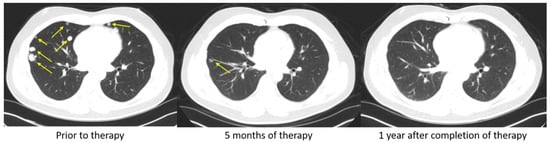
Figure 2 demonstrates a patient with advanced HCC that progressed on sorafenib and subsequently achieved a complete response after one year on nivolumab. In another phase III clinical trial, Finn et al. studied the effects of Atezolizumab, an anti-PD-L1 monoclonal antibody, in combination with Bevacizumab, a vascular endothelial growth factor (VEGF) inhibitor, in advanced HCC. The combination of Atezolizumab with Bevacizumab led to an overall survival of 67.2% at 12 months and a median progression-free survival of 6.8 months. This was superior to sorafenib, which had a 12-month overall survival and median progression-free survival of 54.6% and 4.3 months, respectively. This trial demonstrates that combination therapy with Atezolizumab with Bevacizumab may be superior to monotherapy with sorafenib in patients with advanced HCC [
48].
Figure 2. Complete response to PD-1 immune checkpoint inhibitors. A 48-year-old man with HBV cirrhosis developed a liver mass that was biopsied and found to be HCC. He underwent a laparoscopic partial right hepatectomy. Pathology revealed a poorly differentiated HCC, 3.3 cm in size, and margins were negative. On surveillance, he was found to have developed a rising AFP and to have bilateral lung nodules (Figure), but there was no evidence of recurrent or metastatic disease in the abdomen or pelvis. Biopsy of one of the pulmonary nodules confirmed metastatic disease and he was started on sorafenib. However, his disease progressed. He had no FGFR alteration, so he was started on nivolumab. After only 5 months of therapy, an objective response was seen on CT scan (middle). He completed a full year of therapy, and he remains without evidence of disease one year after completion of therapy.
While these studies demonstrated a moderate improvement in outcomes with the use of specific ICIs as monotherapy compared with sorafenib, the benefits are more obvious when different ICIs are combined. Combination therapies allow targeting of multiple, upregulated immune checkpoint pathways, which may improve patient outcomes versus monotherapy with ICIs. However, these findings must be balanced with the potential for increased toxicity and immune-related adverse events. The value of targeting multiple ICIs has been demonstrated in patients with melanoma, and this strategy is under active investigation in patients with HCC [
49]. One ongoing study (NCT02519348) is treating patients with HCC with durvalumab (anti-CTLA-4) and tremelimumab (anti-PD-1 agent). Early results from this phase I/II trial indicated that a single, increased dose of tremelimumab combined with a standard regimen of durvulumab led to an objective response rate (ORR) of 22.7% and a median overall survival of 18.7 months, with an acceptable side effect profile [
50]. The combination of tremiliumumab and durvulumab is now being studied in an ongoing phase III clinical trial (NCT03298451) as a first-line treatment for advanced HCC in patients who are ineligible for locoregional therapy. Another study (NCT01658878) reported the safety and efficacy of nivolumab in combination with ipilimumab in patients with advanced HCC. The study included three arms with combination therapy, each with a varying dose and schedule of nivolumab and ipilimumab. The lack of comparison to monotherapy arms makes the synergistic benefits unclear; however, the investigators concluded that patients on both medications developed a 31% ORR, which included a complete response rate of 6%, partial response rate of 24%, and a tolerable safety profile [
46]. Another study from the same investigator (NCT01658878) demonstrated that adding ipilimumab to a regimen of nivolumab and cabozantinib (receptor tyrosine kinase inhibitor) resulted in an improved ORR and median progression-free survival, but at the cost of increased grade 3 and 4 adverse events [
51]. In addition to these trials, numerous ongoing clinical trials are being conducted to determine the therapeutic benefit of various ICI combinations [
52]. Other studies from the National Cancer Institute (NCT02465060) and from the American Society of Clinical Oncology (NCT02693535) are using next-generation sequencing of paraffin-embedded tumors after resection to improve patient selection for available systemic therapies. Both studies are investigating a wide variety of cancer types, including hepatobiliary cancers. Genotyping studies such as these will determine the utility of individualized treatment and may eventually inform the use of individualized immunotherapies.
With evidence mounting to support the use of immunotherapy for advanced HCC alone or in combination with other systemic therapies, other studies have been conducted to investigate whether there is an advantage of combining ICIs with liver-directed therapies. A phase I/II study by Duffy et al. combined tremelimumab (anti-CTLA-4) with transarterial chemoembolization (TACE) or ablation and reported a median overall survival of 12.8 months. Notably, patients who received TACE instead of ablation had improved survival, with a 12-month survival rate of 80.8%. The investigators also noted an increase in activated CD4+ and CD8+ T cells in the peripheral blood, though differences in TIL between pre- and post-treatment samples were not significant. In addition, 12 out of 14 patients with concurrent hepatitis C infection had reduced viral loads. While impressive, this early study lacked control arms of patients treated with transarterial therapy alone or immunotherapy alone [
53]. Additional clinical trials (NCT04517227, NCT03753659, and NCT04102098) to evaluate the efficacy of PD-1 inhibitors combined with ablation or TACE for HCC are currently accruing patients. In addition, several other clinical trials are evaluating the value of adding ICIs to other systemic therapies and small-molecule inhibitors such as vascular endothelial growth factor (VEGF) inhibitors and kinase inhibitors [
52]. Such studies include NCT03713593, NCT03605706, and NCT03006926, among others, and are anticipated to complete accrual in the next 2–5 years.
Other ICIs are in development but have yet to see widespread clinical use. Lymphocyte-activation gene 3 (LAG-3) and T-cell immunoglobulin mucin-3 (TIM-3) are being studied as potential targets for ICIs. LAG-3 binds to major histocompatibility complex (MHC) class II and is found on CD8+ T cells [
54]. LAG-3 expression is increased in HCC tumor-infiltrating lymphocytes (TILs) and LAG-3 expression is correlated with impaired T cell effector function [
55]. Mouse models have been used to validate the efficacy of LAG-3 blockade. In mice, anti-PD-1 and anti-LAG-3 antibodies work synergistically to induce a complete response in the majority of those bearing syngeneic melanoma or colorectal tumors. This is in contrast to only a small minority of mice that develop complete remission when treated with either agent alone [
56]. Based on these preclinical results, phase I clinical trials are now open and are evaluating the safety and efficacy of LAG-3-targeted ICIs in HCC. NCT04658147 is evaluating Relatlimab, an anti-LAG-3 monoclonal antibody, among patients with HCC, while NCT03849469 is assessing the utility of a bispecific antibody (an antibody capable of binding two receptors at once) that targets both CTLA-4 and LAG-3.
Table 1 contains a full list of ongoing clinical trials using a combination of immune checkpoint inhibitors and other novel immunotherapies for HCC.
Table 1. Clinical trials currently evaluating immunotherapies in HCC. List of clinical trials involving combination immune checkpoint inhibitors, adoptive cell transfer, and other innovative immunotherapies in HCC. Source:
clinicaltrials.gov (28 July 2021). HCC, hepatocellular carcinoma; TIL, tumor infiltrating lymphocyte; CAR-T cell, chimeric antigen receptor T cell.
| Trial Identifier |
Phase |
Status |
Immunotherapy |
Co-Treatments |
Participants |
| NCT04740307 |
II |
Recruiting |
Combination immune checkpoint inhibitors (CTLA-4 with PD-1) |
Lenvatinib |
110 |
| NCT02821754 |
II |
Recruiting |
Combination immune checkpoint inhibitors (CTLA-4 with PD-1) |
Transarterial catheter chemoembolization, radiofrequency ablation, cryoablation |
90 |
| NCT04785287 |
I/II |
Recruiting |
Combination immune checkpoint inhibitors (CTLA-4 with PD-1) |
Stereotactic Body Radiation Therapy |
80 |
| NCT04430452 |
II |
Not yet recruiting |
Combination immune checkpoint inhibitors (CTLA-4 with PD-1) |
Hypofractionated Radiation Therapy |
30 |
| NCT03638141 |
II |
Recruiting |
Combination immune checkpoint inhibitors (CTLA-4 with PD-1) |
Drug-eluting bead transarterial chemoembolization |
30 |
| NCT03222076 |
II |
Active not recruiting |
Combination immune checkpoint inhibitors (CTLA-4 with PD-1) |
N/A |
30 |
| NCT03652077 |
I |
Active, not recruiting |
Immune checkpoint inhibitor (TIM-3) |
N/A |
40 |
| NCT03680508 |
II |
Recruiting |
Combination immune checkpoint inhibitors (TIM-3 with PD-1) |
N/A |
42 |
| NCT04658147 |
I |
Recruiting |
Combination immune checkpoint inhibitors (LAG-3 with PD-1) |
N/A |
20 |
| NCT03695250 |
I/II |
Active, not recruiting |
Immune checkpoint inhibitor (PD-1) with IDO1 inhibitor |
N/A |
8 |
| NCT04728321 |
II |
Recruiting |
Bispecific antibody against CTLA-4 and PD-1 |
lenvatinib |
75 |
| NCT04444167 |
I/II |
Recruiting |
Bispecific antibody against CTLA-4 and PD-1 |
Lenvatinib |
30 |
| NCT04601610 |
I/II |
Not yet recruiting |
Bispecific antibody against CTLA-4 and PD-L1 |
Ningetinib Tosylate (tyrosine kinase inhibitor) |
70 |
| NCT03849469 |
I |
Recruiting |
Bispecific antibody against CTLA-4 and LAG-3, combination bispecific antibody and immune checkpoint inhibitor (PD-1) |
N/A |
242 |
| NCT03980288 |
I |
Recruiting |
GPC3 CAR-T cell |
Fludarabine, cyclophosphamide |
36 |
| NCT04121273 |
I |
Recruiting |
GPC3 CAR-T cell |
N/A |
20 |
| NCT03884751 |
I |
Recruiting |
GPC3 CAR-T cells |
N/A |
15 |
| NCT02905188 |
I |
Recruiting |
GPC3 CAR-T cells |
Cytoxan, Fludarabine |
14 |
| NCT04951141 |
I |
Recruiting |
GPC3 CAR-T cells |
N/A |
10 |
| NCT03302403 |
N/A |
Active, not recruiting |
GPC3 CAR-T cell |
Fludarabine, Cyclophosphamide |
18 |
| NCT04506983 |
1 |
Not yet recruiting |
GPC3-CAR-T cells |
N/A |
12 |
| NCT03198546 |
I |
Recruiting |
GPC3 and/or TGFβ targeting CAR-T cells |
N/A |
30 |
| NCT03638206 |
I/II |
Recruiting |
DR5 CAR-T cells |
N/A |
73 |
| NCT03941626 |
I/II |
Recruiting |
DR5 CAR-T/TCR-T cells |
N/A |
50 |
| NCT03993743 |
I |
Recruiting |
CD147 CAR-T cells |
N/A |
34 |
| NCT04550663 |
I |
Not yet recruiting |
NKG2dLs CAR-T cells |
N/A |
10 |
| NCT03441100 |
1 |
Recruiting |
MAGEA1 TCR engineered T cells |
Cyclophosphamide, fludarabine, Interleukin-2 |
15 |
| NCT04502082 |
I/II |
Recruiting |
Alpha fetoprotein peptide/HLA-A2 complex TCR engineered T cells |
N/A |
50 |
| NCT04634357 |
I/II |
Not yet recruiting |
Alpha fetoprotein peptide/HLA-A2 complex TCR engineered T cells |
N/A |
25 |
| NCT04518774 |
I |
Recruiting |
expanded allogeneic gamma-delta T cells |
N/A |
8 |
| NCT03836352 |
II |
Recruiting |
Induction of survivin-specific cytotoxic T lymphocytes with immune checkpoint inhibitors (PD-1) |
Cyclophosphamide |
184 |
| NCT04417764 |
I |
Recruiting |
PD-1 knockout engineered T cells |
Transarterial catheter chemoembolization |
10 |
| NCT04317248 |
II |
Recruiting |
Autologous dendritic cell vaccine |
Cyclophosphamide |
600 |
| NCT04912765 |
II |
Recruiting |
Neoantigen Dendritic Cell Vaccine with immune checkpoint inhibitor (PD-1) |
N/A |
60 |
| NCT04147078 |
I |
Recruiting |
Autologous dendritic cells vaccine |
N/A |
80 |
| NCT03228667 |
II |
Active, not recruiting |
PD-L1 targeting high-affinity natural killer with immune checkpoint inhibitors (PD-1 or PD-L1) |
N-803 (interleukin-15 superagonist) |
145 |
| NCT03311334 |
I/II |
Recruiting |
Peptide vaccine against WT1 with immune checkpoint inhibitors (PD-1) |
N/A |
104 |
| NCT04246671 |
I/II |
Recruiting |
Peptide vaccine against HER-2/neu |
N/A |
45 |
| NCT02432963 |
I |
Active, not recruiting |
Peptide vaccine against P53 with immune checkpoint inhibitors (PD-1) |
N/A |
19 |
| NCT04251117 |
I/II |
Recruiting |
Personalized neoantigen DNA vaccine with immune checkpoint inhibitor (PD-1) |
Plasmid-encoded interleukin 12 |
24 |
| NCT04248569 |
I |
Recruiting |
Peptide vaccine against DNAJB1-PRKACA fusion kinase with combination immune checkpoint inhibitors (CTLA-4 with PD-1) |
N/A |
12 |
| NCT03071094 |
I/II |
Active, not recruiting |
Oncolytic vaccine with immune checkpoint inhibitor (PD-1) |
N/A |
30 |
| NCT04665362 |
I |
Not yet recruiting |
Oncolytic vaccine with immune checkpoint inhibitor (PD-1) |
Apatinib |
10 |
| NCT04665362 |
I |
Not yet recruiting |
Oncolytic virus with immune checkpoint inhibitor (PD-1) |
Apatinib |
10 |
TIM-3 is expressed on a variety of cells, including innate immune cells, CD4+ cells, and CD8+ cells [
57,
58]. Similar to LAG-3, PD-1, and CTLA-4, TIM-3 has been demonstrated to impair the adaptive immune response to cancer when bound to its cognate ligand [
59]. TIM-3 is upregulated in the TILs of HCC patients, and increased TIM-3 expression is associated with decreased survival and increased recurrence [
60]. Meanwhile, blocking TIM-3 binding to its ligand, galectin-9, reduces the population of regulatory T cells (T cells that inhibit the immune response to cancer), improves TIL proliferation, and improves effector cytokine production [
58,
61]. These studies highlight the importance of TIM-3 in the immune response to HCC. There are several ongoing phase I and phase II clinical trials that are evaluating the efficacy and safety of TIM-3 ICIs [
59]. One study from the University of Hawaii (NCT03680508) is specifically studying cobolimab, an anti-TIM-3 antibody, with dostarlimab, an anti-PD-1 antibody, in 42 liver cancer patients. While promising, the efficacy of TIM-3 ICIs in humans remains unknown.
The efficacy of immune checkpoint inhibitors may be improved with the targeting of Indoleamine 2,3-dioxigenase 1 (IDO1). IDO1 is involved in the conversion of L-tryptophan into L-kynuernine and promotes immunosuppression by depleting L-tryptophan in effector cells and producing L-kynuernine derivatives that ultimately create signals leading to increased regulatory T cell differentiation [
62,
63]. In mouse models of HCC, IDO1 blockade along with anti-CTLA-4 treatment proved a more effective means to reduce tumor growth than anti-CTLA-4 monotherapy, paving the way for IDO1 inhibitors to be tested in combination therapies [
64]. Currently, one clinical trial (NCT03695250) is investigating the safety and efficacy of an IDO1 inhibitor, BMS-986205, paired with nivolumab in patients with stage III and stage IV HCC.
2.4. Future Directions and Novel Approaches
Other forms of immunotherapy beyond ICIs are currently in development and may be tested in patients with HCC. One such example is the adoptive cell transfer (ACT) of immune cells. ACT involves the collection of immune cells from a cancer patient, ex vivo enrichment and expansion or genetic modification, followed by the administration of the expanded T cells back into the patient [
65]. The ACT of tumor-infiltrating lymphocytes (TIL) has proven to be an effective treatment in advanced melanoma, some types of cervical cancer, and small cell lung cancer, but its interest in HCC is still being investigated [
65,
66,
67]. In an early randomized trial using autologous tumor-reactive peripheral blood mononuclear cells (PBMC) harvested prior to surgical resection, transfer of PBMCs led to an 18% decrease in recurrence compared with corrected control patients that only underwent surgery. However, with a median follow-up of 4.4 years, overall survival was no different between the two cohorts [
68].
Several studies have investigated the use of another type of cellular therapy in patients with HCC called cytokine-induced killer (CIK). CIK cells are expanded ex vivo from peripheral blood mononuclear cells and function to improve the immune response to cancer through non-MHC-restricted antitumor activity. In a meta-analysis of 13 clinical trials, CIK cell infusion was noted to improve 1- and 2-year survival in patients with HCC [
69]. In this study, Pan et al. identified independent predictors of overall survival in CIK immunotherapy, including tumor size, tumor capsule, pathological grades, total bilirubin, albumin, prothrombin time, alpha-fetoprotein, and tumor number [
70,
71]. While the results of CIK treatments for HCC have been promising, and large-scale, phase III studies have been performed, the treatment has yet to be widely implemented.
More recently, CAR-T immunotherapy, which genetically modifies T cells using lentiviruses or retroviruses, has emerged as a promising therapy. These T cells begin to express CAR, which can bind antigens without MHC antigen presentation. Once CAR-T cells are identified, they are expanded ex vivo and these cells are adoptively transferred into the patient [
72,
73]. Due to the harsh tumor immune microenvironment, the use of the adoptive transfer of ex-vivo-expanded tumor reactive T cells has gained traction. By enriching for a specific population of T cells that have a known affinity for a patient’s cancer-specific antigens and injecting these cells at large numbers into the patient, researchers have tried to overcome the harsh immune-inhibitory tumor microenvironment and induce tumor regression. In xenograft models of HCC, CAR-T cells have been used successfully to inhibit tumor growth and, in some instances, eradicate tumors. However, concerns exist about the safety of CAR-T immunotherapy due to the potential for off-target toxicity, as well as their ability to effectively function in the immunosuppressive environment of the human liver [
74,
75,
76]. The future of adoptive cell transfer for HCC will likely require more studies. Fortunately, numerous ongoing clinical trials are investigating the utility of ACT for HCC with a particular emphasis on CAR-T immunotherapy.
Inducing an immune response to cancer through the administration of tumor surface antigens that are shared between tumors and patients is another approach currently being investigated. Tumor peptides can be packaged or delivered in various ways to optimize the immune response. Cancer vaccines are based on the delivery of full-length tumor antigens or corresponding epitope peptides. These antigenic sequences can be delivered either as naked peptides/proteins, naked DNA/RNA, or vectored via recombinant viruses (oncolytic or not) or even bacteria, or via loaded cells such as dendritic cells. One phase I clinical trial in HCC patients comparing dendritic cell infusion, dendritic cell infusion with sorafenib, and sorafenib alone noted that dendritic cell infusion led to increased tumor-specific CD8+ populations in peripheral blood, without evidence of autoimmune reactions, and with a disease control rate of 35%. Overall survival was difficult to assess as median overall survival was much lower than that in historical controls [
77].
Traditional peptide vaccines have also been studied. A glypican-3-derived peptide vaccine was well tolerated in a phase I trial. Out of 33 patients treated, only one partial response was observed; nineteen patients had stable disease and the remaining had progressive disease. The median overall survival in this study was 9 months [
78]. Another peptide vaccine, this time with a telomerase peptide, did not exhibit antitumor efficacy in a phase II clinical trial of HCC patients [
79]. Numerous other peptide vaccine candidates are being studied in HCC, including alpha-fetoprotein, NY-ESO, SSX-2, and melanoma antigen-encoding gene A [
80]. Peptides can also be packaged into viruses that are designed to infect rapidly dividing cancer cells. After tumor cell infection, viral RNAs are expressed and encode for antigenic peptide or protein sequences, thus aiding in stimulating an immune response to cancer. This exciting approach has been tested clinically in patients with HCC. Pexastimogene devacirepvec (Pexa-Vec), an oncolytic vaccine that preferentially infects HCC cancer cells, increased overall survival, particularly when administered at high doses (109 PFU), in a phase II clinical trial [
81]. A phase III clinical trial, NCT02562755, evaluating the efficacy of Pexa-Vec, was completed in December 2020 and is awaiting published results. The results of these promising studies may determine the direction of novel immunotherapies in HCC.
While progress is being made and immunotherapy is proving itself to be an important approach in the armamentarium of the treating oncologist, significant challenges remain. Unlike other tumors, such as pancreatic cancer, colon cancer, or malignant melanoma, there is no reliable biomarker available to predict tumor response to immunotherapy in patients with HCC [
82,
83]. In addition, the fact that neither tumor mutational burden nor the expression of PDL-1 is predictive of the tumor response to ICIs in patients with HCC makes patient selection for immunotherapy difficult (Oncotarget 2019, 10, 4018–4025; Genome Med. 2019, 11, 28). Despite these challenges, novel clinical trials and continued efforts will ultimately decide the continued role of immunotherapy in patients with HCC.