Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, General & Internal
Nitric Oxide (NO) is a potent signaling molecule involved in the regulation of various cellular mechanisms and pathways under normal and pathological conditions. NO production, its effects, and its efficacy, are extremely sensitive to aging-related changes in the cells.
- nitric oxide
- nitric oxide synthase
- aging
- senescence
- NO signaling pathways
- therapeutic agents
1. Introduction
Nitric oxide (NO) is one of the main signaling molecules in the body that shows its principal performance in an unconventional manner. NO exerts its effects on several molecular targets and can regulate various functions such as neurotransmission, vascular tone, transcription of genes, translation of mRNA, and protein post-translational modifications [1]. NO can react with superoxide anion (O2−), resulting in the formation of potent oxidant peroxynitrite (ONOO−), and subsequently interacting with biomolecules such as proteins, lipids, and DNA via direct oxidation reactions or indirect radical-mediated mechanisms [2][3][4][5][6]. ONOO− is responsible for many pathological processes in mammalian organelles including cytotoxicity induction, oxidation, protein modifications, lipids peroxidation, DNA damage, cell death, mitochondrial disruption, dysregulation of signal transduction, and apoptosis [7]. Besides, various methods are available for the synthesis of ONOO−. ONOO−, as a highly reactive molecule, can result in the generation of oxidizing and nitrating species [6]. ONOO− and its decomposition yields containing NO2, CO−, and OH can impair several reactions comprising the tyrosine nitration of proteins, the inactivation of superoxide dismutase (SOD), and tissue damage [8]. ONOO− induced nitrosative stress has the capacity to induce the appearance of breaks in single-strand DNA, which subsequently activates the poly-ADP-ribose polymerase (PARP) [9]. NO is produced in mammals by three distinct forms of NO synthase (NOS), coded by three distinct genes: neuronal ‘n’NOS (or NOS-I), inducible ‘i’NOS (or NOS-II), and endothelial ‘e’NOS (or NOS-III). All NOS proteins are homodimers [10][11][12]. Furthermore, in mammals, NO can also be formed, resulting in NOS-independent pathways, explicitly by consecutive reduction of nitrate (NO3−) and nitrite (NO2−). NO2− has the capability to be univalently reduced to NO through the transition of metal-comprising enzymes, e.g., deoxymyoglobin/deoxyhemoglobin (deoxyHb/deoxyMb), and xanthine oxidase (XO) at the time in which the partial pressure of oxygen (pO2) levels is low. The aforementioned NOS-independent gates for NO production represent the NO3−/NO2−/NO pathway or O2-independent formation of NO [13]. NO signaling, NO donors, and NOS inhibitors are very important in the pathophysiology of age-related diseases and their associated putative therapeutic approaches. In this study, we review the role of NO in the aging processes of cells.
2. Mechanisms of NOS
The common substrate for all NOS enzymes is l-arginine. NOS enzymes simultaneously bind multiple cofactors and prosthetic groups: heme, glutathione, molecular oxygen, reduced nicotinamide-adenine-dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), flavin mononucleotide (FMN), (6r-)-tetrahydro-l-biopterin (BH4), and Ca2+ calmodulin [1]. NOS enzyme uses the flavins FAD and FMN to transfer the electrons from the carboxy-terminal reductase domain of NADPH to the amino-terminal oxygenase domain of heme. The transferred electrons are used to reduce and activate O2 and oxidize l-arginine to l-citrulline and NO. NOS catalyzes two-step oxidation of l-arginine: firstly, l-arginine is hydroxylated to N-hydroxy-l-arginine, which subsequently is oxidized to l-citrulline and NO [14][15]. Binding sites of BH4 and l-arginine are located at the cysteine ligand of the heme and the oxygenase domain. The latter is the binding site for molecular oxygens as well [16][17].
The process of electron transfer from NADPH to the heme in nNOS and eNOS is facilitated by the binding of calmodulin. The elevation of intracellular Ca2+ activity is a key factor responsible for the increased affinity of calmodulin to NOS. However, even at low levels of intracellular Ca2+ concentrations, calmodulin-binding with iNOS does not change because of the distinct amino acid composition of the calmodulin-binding site [18][19][20]. NO affects several enzymes and proteins, including the activation of soluble guanylyl cyclase and the production of cyclic guanosine monophosphate (cGMP) [1]. Regulation of the function of smooth muscle by cGMP, which is associated with the mediation of phosphodiesterase (PDE) isozymes, plays a part in smooth muscle relaxation [21]. NO production pathways and NO functions are shown in (Figure 1).
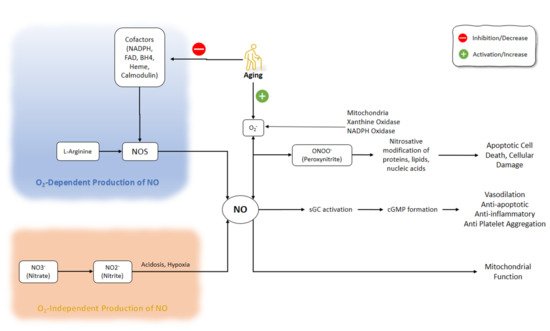
Figure 1. NO production pathways and NO functions. There are several critical steps in the production of NO that might be O2-dependent or O2-independent. Inhibition of NOS cofactors is important in the aging process. Besides, aging is accompanied by the activation of superoxide. NO: Nitric Oxide; NOS: Nitric Oxide Synthase; NADPH: nicotinamide-adenine-dinucleotide phosphate; FAD: flavin adenine dinucleotide; BH4: (6r-)-tetrahydro-l-biopterin.
2.1. nNOS
Ca2+ and calmodulin control neuronal NOS (nNOS) activity. The subcellular distribution and function of nNOS were determined by the direct association of the PDZ domain of nNOS with other proteins [22]. PDZ domain brings up to an area that entails 80–120 amino acid residues that perform as components taking part in protein-protein interactions [22][23]. The canonical PDZ domain of nNOS connects to the nNOS adaptor protein NOS1AP, also well known as carboxy-terminal PSD-95-Dlg-ZO1 [PDZ] ligand of nNOS (CAPON). The ternary complex made by PDZ domain interactions between N-methyl-d-aspartate (NMDA) receptor, PSD-95, and nNOS performs the action of scaffolding nNOS to the NMDA receptor, and NMDA-encouraged Ca2+ influx is consequently well coupled to the activation of nNOS and the subsequent formation of NO [24]. The nNOS has been found in various parts and tissues of the body, including the brain, spinal cord, sympathetic ganglia, adrenal glands, peripheral nitrergic nerves of the nervous system, as well as epithelial cells of different tissues such as kidney macula densa cells, pancreatic islet cells, and vascular smooth muscle cells. The most abundant presence of nNOS has been determined in the mammal skeletal muscle [25].
nNOS does not seem to be involved in the acute neurotransmission in the central nervous system (CNS); however, it can mediate the long-term regulation of synaptic transmission (long-term potentiation, long-term inhibition). Furthermore, nNOS is involved in the central regulation of blood pressure. This has been shown by the induction of systemic hypertension while blocking nNOS activity in the medulla and hypothalamus [1]. The effects of nNOS on the regulation of vascular tone are independent of its CNS effects. The production of NO by nerves that contain nNOS (nitrergic nerves) stimulates NO-sensitive guanylyl cyclase and decreases the tone of the innervated smooth muscles in the periphery including blood vessels [25][26]. Ca2+/calmodulin (CaM)-dependent protein kinases, cyclic AMP-dependent protein kinase (PKA), cGMP-dependent protein kinase, and protein kinase C (PKC) participate in the phosphorylation of nNOS [27].
The effects on the smooth muscles of the corpus cavernosum and the relaxation of nitrergic nerves affects penile erection [28][29]. The relaxation of these smooth muscle cells is mediated by cyclic GMP, which can be degraded by phosphodiesterases (predominantly isoform 5). Therefore, a residual nNOS activity is needed for the selective PDE5 inhibitors to be effective [30].
2.2. iNOS
iNOS enzymes are exclusively located in macrophages and their production is triggered when needed. The bacterial lipopolysaccharides and cytokines can influence the expression of iNOS [25][31]. When iNOS is expressed, it is activated continuously and is not affected by the intracellular Ca2+ concentrations. The iNOS in macrophages excretes large amounts of NO, which is important for them as a toxic defense molecule [32]. NO can disrupt the enzymes containing iron in their catalytic centers, such as iron-sulfur cluster-dependent enzymes (complexes I and II) entailed in mitochondrial electron transport, ribonucleotide reductase (the rate-limiting enzyme in DNA replication), and cis-aconitase (a key enzyme in the citric acid cycle) [32][33]. Recently, studies on 13C tracing and mitochondrial respiration revealed a new observation on the modulation in NO-mediated macrophage metabolic programming attributed to the regulation of the tricarboxylic acid (TCA) cycle and the accumulation of itaconate with a great fractional combination of glucose carbon at the overhead of glutamine carbon, which could link the suppression of pyruvate dehydrogenase (PDH) and aconitase to NO. NO has a functional impact on the accumulation of metabolite hallmarks of M1 polarization, including citrate, succinate, and itaconate. Indeed, during the stimulus, glucose uptake was elevated, immune-response gene-1 (Irg1) was upregulated, and citrate and itaconate were produced, incorporating glucose-derived carbon. [34][35][36]. At higher concentrations, NO can directly impede target cell DNA and affect strand breaks and fragmentation [37][38]. In addition to macrophages, other non-immune cells can produce NO by iNOS upon induction via cytokines and thus can affect other cells, i.e., endothelial cells exert NO-mediated lysing of tumor cells and cytokine-induced iNOS in hepatocytes synthesize NO to kill malaria sporozoites [39][40].
2.3. eNOS
eNOS is the leading source of NO in vascular endothelium. Its promotor expression exists in various types of cells such as cardiomyocytes, platelets, specific brain neurons, in syncytiotrophoblasts of the human placenta, and in LLC-PK1 kidney tubular epithelial cells [1][25]. NO production is regulated by Ca2+-activated calmodulin as the increase of intracellular Ca2+ induced binding of calmodulin subsequently increases the eNOS activity [19]. Other factors can also regulate the activity of eNOS, e.g., heat shock protein 90 (hsp90) acts as an allosteric modulator that promotes the eNOS’ (re)coupling and therefore its activity [41][42]. Another regulator of eNOS is caveolin-1 [43]. It is a tonic inhibitor interacting with the eNOS localized in caveolae [44]. The lack of caveolin-1 results in enhanced endothelium-dependent relaxation in mice [45]. The inhibitory effect of caveolin-1 can be blocked by the presence of calmodulin and hsp90 [46]. Fluid shear stress can activate eNOS independent of intracellular Ca2+ increase through phosphorylation of eNOS at several serine (Ser), threonine (Thr), and tyrosine (Tyr) residues [47][48]. During the regulation of eNOS activity by phosphorylation, the probable changes in phosphatase activity are able to have an impact on NO generation. Certainly, protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) play diverse roles in the regulation of eNOS phosphorylation [48]. According to recent studies, aging is considered to be related to the greater protein acetylation levels and the defeat of sirtuin-1 deacetylation activity, which could subsequently cause the reduced eNOS activity [49][50]. Four lysine residues are found within the cAMP-response element-binding protein (CREB)-binding protein (CBP) of eNOS. The identification of two of these residues as a consequence of the acetylation suggests that SIRT1 may target more than a single residue within this domain, regulating enzymatic activity via the modulation of deacetylation of the targeted residues. Moreover, by knowing the recognized role of the phosphorylation in eNOS regulation, in a similar manner to other proteins, the acetylation and phosphorylation might both be responsible for the regulation of eNOS activity [51].
3. Aging and NO Signaling
Aging can negatively affect cellular functions even in the absence of obvious disease. However, the disorders emerge with aging in several systems and organs including the cardiovascular system, CNS, skin, kidneys, thyroid, male and female reproductive system, muscles, and immune system. The underlying mechanisms of these age-related disorders are complex and involve multiple pathways, factors, and cellular targets which are beyond the scope of this review. Herein, we made an in-depth focus on the involvement of NO signaling pathways in the complex scenario of aging-related disorders. The disturbed NO signaling contributes to a variety of aging pathologies. In a broad sample (763 participants, age range of 19–107 years), Montesanto et al. (2013) explored the genetic diversity in human NOS genes and found the association between decreased cognitive function and physical performance with nNOS rs1879417 and iNOS rs2297518 polymorphism, respectively [52].
3.1. Cardiovascular Aging and NO
The impairment of the vascular function due to endothelial dysfunction is one of the main heart and vasculature alterations during cardiovascular aging. The endothelial dysfunction is characterized by the impaired vasodilator reaction to the flow or the agonists and one of its fundamental causes is the decreased bioavailability of NO under pathological conditions such as atherosclerosis, hypertension, and hypercholesterolemia [53][54][55][56]. The reduced abundance or activity of eNOS, the increased levels of endogenous NOS inhibitors, and the reduced supply of l-arginine, or the increased NO degradation or scavenging, can result in decreased NO bioavailability.
As part of the urea cycle, hydrolyzing l-arginine to ornithine and urea by arginase can affect the bioavailability of l-arginine. NO performance increases through the iNOS in the macrophages following the inhibition of arginase [57]. Arginase can also modulate the vascular tone by regulating l-arginine supply for eNOS activity. Berkowitz et al. (2003) have shown that arginase activity and expression are upregulated in the aorta rings of old rats while the NOS activity and cGMP levels and, consequently, the endothelial activity, were reduced. Moreover, arginase inhibition increased vasodilation in the aortic rings of old rats compared with young adult rats. Furthermore, arginase inhibition restored NOS activity and cyclic GMP levels in the old rat vessels compared with those of young rats. Arginase competitively controlled the bioavailability of l-arginine for eNOS by limited NO production despite the increased expression of eNOS or adding exogenous l-arginine. The pretreatment with arginase inhibitors was sufficient to restore l-arginine responsiveness in the old rat vascular ring preparations. This result indicates that the enzyme kinetics strongly favors arginase. During arginase inhibition, l-arginine can restore cytosolic NOS responsiveness and thus restore vasorelaxation. Arginase was in contact with NOS, contributing to impaired relaxation of the endothelium. In brief, Berkowitz et al.’s (2003) research showed that arginase expression and activity increases in the vasculature with advancing age and leads to endothelial dysfunction [58].
Chou et al. (1998) found lower expression and activity of eNOS in spontaneously hypertensive rats (SHR) compared with non-SHR at both young and old age. The SHR developed the early onset of the decrease in eNOS expression and activity. However, the eNOS decline did not progress by aging in a way that eNOS activity was lower in the old non-SHR rats compared to the old SHR rats. The iNOS expression and activity showed inversed correlation to eNOS in both SHR and non-SHR. The iNOS expression was increased further by lipopolysaccharide (LPS) stimulation only in the 14–17- and 63-week groups of SHR. Hypertension control could regulate the abnormal iNOS expression and activity in SHR but, a “cause and effect” relationship was not established. Besides, the increase in levels of the tumor necrosis factor-alpha (TNF-α) and NO2−/NO3− induced by LPS was much higher in SHR than in non-SHR at both groups of 14–17- and 63-week [59]. In the studies of Wu et al. (1999), the significant elevation was observed in basal plasma and aorta levels of NO2− in SHR treated with LPS model. In vitro, the ACh-induced relaxation was decreased in the aortae isolated from SHR. However, this dissimilarity between the SHR and non-SHR was eliminated after the treatment of the rings with nitro-l-arginine methyl ester (l-NAME), and the damage of NO production was observed in the SHR. Expression of iNOS in both groups treated with LPS was augmented. These data supported the hypothesis that the raised plasma NO level in SHR might be the result of the NO release from protein-bound dinitrosyl nonheme iron complexes (DNIC) in the vascular bed, leading to the abolished hypertension [60]. Vaziri et al. (1998) concluded that the l-arginine/NO pathway was upregulated in young SHR before and after the onset of hypertension, which showed that NO formation was augmented in young SHR both before and after the commencement of the hypertension [61]. Further studies have demonstrated that the elevated production of NO in SHR, which was additionally improved by LPS treatment, is related to a primary expression of iNOS [62].
Cernadas et al. (1998) showed that aged rats had an abnormal hypotensive response to acetylcholine and bradykinin and a retained hypotensive response to nitroprusside sodium. Furthermore, the pressure effect of l-arginine antagonist (l-Nv-nitro-l-arginine) caused an increased sensitivity response and angiotensin II caused a decreased vasoconstrictor response in aged rats. The latter was improved after inhibition of NOS. Aged rats had higher plasma levels of nitrite and nitrate, higher cGMP content, and elevated eNOS and iNOS expression in the aortas. However, the activity of eNOS was significantly decreased in aged rats compared to the young ones, these results correlated with the iNOS activity. The locally released cytokines from endothelial injury sites in the vascular wall might have induced iNOS expression. The iNOS-related NO could decrease the action of the eNOS, thereby favoring impaired endothelium-dependent vasorelaxation. On the other hand, during aging, iNOS activity could play a significant function in retaining the vascular tone [63].
The reduction in endothelial vasoreactivity and excessive diastolic relaxation is linked with cardiovascular aging. Zeiman et al. (2001) showed that l-arginine increased ventricular relaxation in both young and aged rat hearts. The increased myocardial NOS-cGMP signaling was correlated with the elevated levels of eNOS protein in cardiac endothelial cells of aged rats. Furthermore, aged rats had elevated calcium-dependent NOS activity. However, in isolated myocytes, there was no difference between NOS activity and protein abundance. Administrating l-arginine decreased baseline isovolumic relaxation and left ventricular end-diastolic strain in both young and aged rat hearts. Sodium nitroprusside (a NO donor) promoted the NO/cGMP mediating pathway, which was attenuated by the soluble guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one [64]. In the studies of the role of soluble guanylyl cyclase (sGC) in aging, it was found that sGC was a heterodimeric enzyme composed of one α1 and β1 subunit that shared a molecule of heme. It was observed that the existence of heme depended on the expression of enzymatic activity and rendered sGC sensitivity to NO. In numerous examples, the activation of sGC and production of cGMP mediated the impact of NO. Moreover, in the vasculature, NO-induced relaxation and repression of platelet aggregation happened through cGMP [65].
Regarding the preclinical findings, asymmetric dimethylarginine (ADMA) is an important endogenous NOS inhibitor in aging-associated cardiovascular risk. In aging-related biological conditions such as telomere shortening and cell senescence, ADMA is implicated [66]. Concentrations of vitamin D have reverse interactions with ADMA concentrations while h-sensitivity C-reactive protein (hs-CRP) has direct interactions with ADMA concentrations [67]. In a study by Scalera et al. (2006), it was observed that l-arginine could avert the commencement of endothelial aging in ADMA or the homocysteine-treated cells’ elevation of the production of NO and, subsequently, the induction of heme oxygenase (HO-1) enzyme activity and protein expression [68].
Vascular endothelial cells produce and release endothelin-1 (ET-1), a potent vasoconstrictor protein [69]. In stable, older, and sedentary adult humans, ET-1 signaling was enhanced, which can cause tonic vasoconstriction in peripheral arteries [70][71][72]. Furthermore, aortic endothelial cells of older individuals have shown a higher synthesis of ET-1 relative to young adult donors with distinct pathologies [73].
Donato et al. (2009) found an inverse relationship between decreased endothelium-dependent dilation and increased expression of the endothelial cell ET-1 protein. Moreover, the endothelium-dependent dilation was suppressed by the ET-1 signaling in old mice compared to the young. In endothelial cells collected from brachial arteries and peripheral veins of healthy young and old men, reduction in neither eNOS nor activated serine 1177 phosphorylated isoform (PeNOS) of eNOS was found. These data suggested the contribution of ET-1 expression and bioavailability, but not eNOS, to the vascular endothelial dysfunction in aging [74].
In vessels of aged animals, the decreased flow-induced dilation mainly depended on the compromised NO-mediated dilation [75][76]. Superoxide (O2−) increases within the vascular wall with aging, which decreases the bioavailability of NO, leading to endothelial dysfunction in aging [76][77][78]. Production of superoxide could increase the interruption of NO resulting in considerable vasomotor dysfunction, which was attributed to vascular aging [79][80]. Collectively, it was proposed that reaction of NO with O2− led to accelerate the production of ONOO−, which attenuated the bioavailability of NO. Moreover, dysregulation of the eNOS caused impairment in the release of NO in the endothelium and increased superoxide formation [81]. This process of aging in the vascular wall can be worsened further as O2− formation can also be improved by a decline in SOD function [82]. Sun et al. (2004) showed a decrease in the shear stress-induced dilation and NO release following aging in rat mesenteric arterioles and arteries. Moreover, the shear stress stimulation of NO synthesis was decreased in aged vessels, evidenced by the decrease in the absolute perfused nitrite (NO2−/NO3−) compared to the young vessels. Furthermore, the aged rats’ vasculature had lower SOD levels due to the decreased extracellular superoxide dismutase (ecSOD) expression, but not the Cu/Zn-SOD and Mn-SOD expression. Suppressed ECSOD expression further decreased the NO bioavailability following the response to the shear stress in the aged vessels. In summary, isolated mesenteric arteries have decreased NO-mediated vasodilation response to the shear stress due to the lower NO bioavailability following the increased O2− production. The latter is caused by the suppressed ECSOD expression and consequently the lower SOD activities [83].
Caveolin, the intrinsic protein of caveolae, is quantitatively linked with the eNOS in ventricular myocytes and endothelial cells [84]. In the former, eNOS is linked with caveolin-3 and, to a lesser degree, caveolin-1, while, in endothelial cells, eNOS is associated only with caveolin-1 [85]. Arreche et al. (2012) found increased NOS activity, decreased eNOS, and caveolin-1 protein levels, as well as increased iNOS activity in the ventricle of the middle-aged rats [86]. The dissociation of caveolin-1 from eNOS correlated with aging. During the hemorrhage-induced hypovolemia, NOS activity and protein levels were increased in the myocardium while the caveolin-1 presence was decreased, which resulted in further dissociation of eNOS. The iNOS increased following blood loss in middle-aged rats. Thus, the modulatory roles of caveolin-1 on cardiac NOS have been demonstrated in the hypovolemic and aging states.
NO contributes to the regulation of water and electrolyte homeostasis in the cardiovascular system [87]. Arza et al. (2015) found that, following 1-month water restriction, both young and adult rats showed decreased NOS expression and activity related to the increased caveolin-1 levels. Interestingly, aging caused functional alterations in the cardiovascular system, evidenced by more remarkable ventricular NOS formation following water restriction and hypovolemia in adult rats. These findings demonstrate the roles of NO and its caveolin regulatory proteins in preserving cardiac physiological function during dehydration and aging [88].
Aging impairs the control of glucose metabolism, resulting in the decreased glucose tolerance and chronic hyperglycemia in the elderly. These changes can mimic accelerated aging and act as a risk factor for cardiovascular diseases [89][90][91]. Levels of NO and peroxynitrite might play a pivotal part in the progression of late impairment in the diabetic vasculature and kidney [92]. NO donor agents and the NO generated by iNOS persuaded insulin resistance via the S-nitrosation of proteins entailed in the primary stages of insulin action, for instance, insulin receptor β (IRβ), insulin receptor substrate 1 (IRS-1), and protein kinase B (Akt). Even though a number of studies have proposed that NO and iNOS might be important in numerous aging-associated complications, the contribution of NO pathways on aging-induced insulin resistance is indistinct [93]. Moreover, hyperglycemia along with aging correlated with endothelial dysfunction, vascular stiffening, and remodeling [94][95]. The basal activity of eNOS and endothelial dysfunction are evident at an advanced age and during hyperglycemia [96][97][98]. In addition, hyperglycemia has been shown to increase iNOS expression and activity, which correlates with excessive NO production and subsequent cellular damage [99].
Rogers et al. (2013) showed increased senescence markers following the glucose level fluctuations in human umbilical vein endothelial cells (HUVECs). The increase of aging markers was due to the elevated Akt’s transcription and expression and its downstream signaling targets. Moreover, eNOS production was decreased, which resulted in iNOS overexpression, decreased bioavailability of NO, and endothelial dysfunction [100].
Yu et al. (2011) reviewed the relationship between aging-induced heart failure and insulin resistance. The precise mechanism was not deceptive in this issue. The documented outcomes showed that insulin resistance might have adverse effects on bioenergetics and myocardial metabolism. Regarding the investigation of isolated perfused hearts obtained from aged rats and cardiomyocytes, the attenuation of contractile reaction and uptake of glucose to insulin, as well as the dampening of the Akt–eNOS–NO pathway due to the post-insulin receptor signaling, were perceived [101].
Classical pro-atherosclerotic stimuli such as oxidized low-density lipoprotein (oxLDL), proinflammatory cytokines, and reactive oxygen species (ROS) caused endothelial cell apoptosis and disrupted the endothelial monolayer integrity leading to vascular damage and atherosclerosis [102][103][104]. The endothelial NO production could inhibit apoptosis by suppressing caspases (a vital factor in apoptosis cascade) through the S-nitrosylation of its critical cysteine [105][106][107]. Hoffmann et al. (2001) reported a three-fold increase in levels of oxLDL, tumor necrosis factor-a-induced apoptosis, and caspase-3-like activity in aged HUVECs compared to the young cells. The substantially suppressed eNOS expression and decreased overall S-NO content implied that eNOS downregulation in aged HUVECs can lead to an age-dependent increase in vulnerability to apoptosis. This was confirmed as the knockdown of eNOS could elevate the apoptosis rate which was abolished by exogenous NO donors. Furthermore, the shear stress did not affect eNOS protein expression, S-NO content, and apoptosis rate in the aged HUVECs. Overexpression of wild-type eNOS restored the antiapoptotic effects of the shear stress but did not affect it when the shear stress was absent. Strikingly, apoptosis in aged HUVECs was further abrogated by the transfection of constitutively active phosphomimetic eNOS (S1177D) [108].
Oelze et al. (2104) showed that, in the absence of glutathione peroxidase-1 (GPx-1), the aging-associated production of oxidants, the activation of the redox system, and the presence of the dysfunctional and uncoupled eNOS are intensified. They found the increased phosphorylation of protein kinase C, protein tyrosine kinase, and eNOS S-glutathionylation associated with an uncoupling in aged peroxidase-1-deficient (GPx-1−/−) mice. The emerging role of GPx-1 ablation in aging animals has a considerable influence on the burden of oxidative damage, representing the experiential variation between vascular dysfunction in aged wild-type and GPx-1−/− mice [109]. GPx-1 is an intracellular antioxidant enzyme which has an important role in the reduction of H2O2 to H2O in order to delimitate its adverse impacts. GPx-1 is a selenocysteine-comprising enzyme that has been involved in the prevention and progression of numerous usual, chronic, and complicated diseases containing cancer, diabetes, and cardiovascular disease [110][111].
Sirtuins are a family of protein deacetylases that regulate aging processes. They depend on NAD+ and thus control cell metabolism along with other cellular functions. Sirtuin 3 (SIRT3) acts mainly by regulating mitochondrial bioenergetics, an essential prerequisite for healthy aging [112][113]. Lu et al. (2020) found that SIRT3 mRNA and protein are decreased in aged human and rat veins. Ad-SIRT3 gene transfer increased the expression and concentration of SIRT3, MnSOD, catalase (CAT), eNOS, and NO. Furthermore, it reduced the neointimal thickness and neointimal area/media area ratio [114]. Mattagajasingh et al. (2007) showed that SIRT1 contributed to the endothelium-dependent vasodilation by activating eNOS in the endothelium of rat aortic rings through deacetylation in a caloric restriction animal model. SIRT1 targeted lysines 496 and 506 in the calmodulin-binding domain of eNOS. Moreover, SIRT1 inhibition decreased NO bioavailability and thus endothelium-dependent vasodilation in arteries [51].
Red blood cells (RBCs) affect NO generation and scavenging by hemoglobin-dependent nitrite. The changes in levels of NO2− could regulate the NO hemostasis as it can be reduced to NO and also can be oxidated into NO3− [115]. RBCs can affect vascular activity by modulating NO through suppressing NO2− oxidation by their antioxidants [116][117]. It has been shown that aged RBCs have lower antioxidant levels [118]. In a study by Owusu et al. (2013), regulatory roles of RBC on vascular hemostasis of NO vasodilatory mechanisms were investigated. Older RBCs had higher NO2-oxidation kinetics and NO scavenging levels compared to young RBCs. The latter was reflected in the inhibited acetylcholine and NO-donor dependent vasodilation in isolated aortic rings. The similarity of NO2− reduction levels between young and old RBCs was confirmed by the inhibition of nitrite-dependent vasodilatation under oxygenated and deoxygenated conditions in old RBCs [119] (Figure 2).
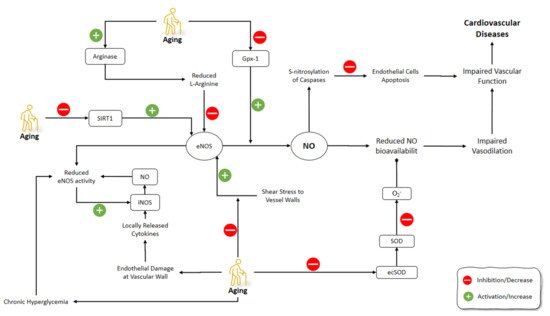
Figure 2. Effects of aging on NO pathways involved in the cardiovascular system. NO: Nitric Oxide; iNOS: Inducible Nitric Oxide Synthase; eNOS: Endothelial NOS; GPx-1: Glutathione peroxidase-1; ecSOD: Extracellular superoxide dismutase; SOD: superoxide dismutase.
3.2. CNS Aging and NO
The brain undergoes morphological and functional alterations during aging. The burden of oxidative stress on the CNS is more prominent due to the relative deficiency in the antioxidant defense mechanisms of CNS [120]. CNS aging is greatly affected by oxidative stress, which involves the production of free radicals including superoxide (O2−), hydrogen peroxide (H2O2), NO, and peroxynitrite (ONOO−) [121][122]. Reactive oxygen species (ROS) containing O2−, H2O2, and the OH−, and reactive nitrogen species (RNS) including ONOO− and NO, are very important in the pathophysiology of neurodegenerative diseases. Due to the great reactive impact of ROS, they chemically interact with various biological molecules, causing significant alterations in cellular tasks, an increased probability of spreading to regions with high metal levels, and induced neuronal death [123][124][125]. Several functions of the CNS are related to NO, including neurotransmitter release, synaptic plasticity, and the regulation of neuronal electrical activity [126][127]. It has been shown that NO has a prominent role in aging processes and related disorders of CNS [128][129] (Figure 3).
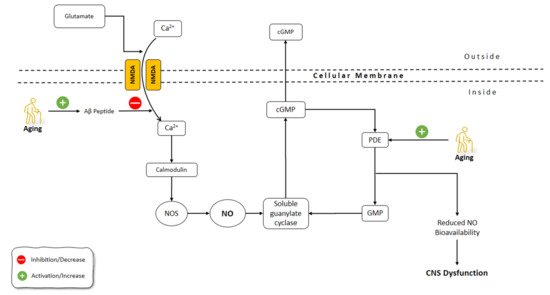
Figure 3. Effects of aging on NO pathways involved in CNS functions. NO: Nitric Oxide; NOS: Nitric Oxide Synthase; Aβ: Amyloid-beta; GMP: guanosine monophosphate; cGMP: Cyclic guanosine monophosphate; Pde: phosphodiesterase; CNS: Central Nervous System; NMDA: N-methyl-d-aspartate receptor.
Uttenthal et al. (1998) observed that in the cortex of the aged rats, nNOS-immunoreactive multipolar neurons were similar to those of the young rats in the terms of quantity, but had an abnormal morphology: varicose, vacuolated, and fragmented structure with an irregular outline and loss of spines. In addition, neurons that were weakly nNOS-positive with a ring of immunoreactive cytoplasm could be found only in the aged rat cortex. On the other hand, iNOS-immunopositive neurons were found only in the aged rat cortex. The old rat cortex had lower nitrotyrosine-positive cells (a biomarker of oxidative stress) compared to the total NOS-positive neurons, while the opposite was observed in the young rat cortex. The formation of nitrotyrosine-containing tissue proteins has the potential to modulate NO and O2− concentrations related to the introduction of protein tyrosyl residues to reaction. Other significant elements that might contribute to the greater protein nitrotyrosine levels identified could be an augmented production of O2− and an abridged rate of turnover of the related tissue proteins with age. According to the outcomes obtained in the study, the increase in nNOS and iNOS expression in the aged rat cortex is not essentially related to the proportional increase in NOS [130]. In a study by Hilbig et al. (2002), a decrease of nNOS expression was observed during the aging process in rats [131].
Brain aging has been linked with mitochondrial dysfunctions [132]. According to recent studies, NOS is not present in the mitochondrial matrix but rather makes part of the outer membrane. Lores-Arnaiz and Bustamante (2011) found a decrease in mitochondrial NO production in both synaptic and non-synaptic neurons of the aged rat cortex. Furthermore, the impairment of mitochondrial functions: ATP deficiency, H2O2 production at high levels, abnormal calcium homeostasis, and decreased levels of NO were observed in the aged rat compared to the young rat cortex neurons [133].
Synaptic mitochondria, which need to sustain the energy required for synaptic activity, showed present functional differences when compared to non-synaptic mitochondria [134]. However, although the aging-induced changes in the brain are thought to be mediated by the increased activity of NOS [135], the experimental data is rather controversial, as the demonstrated results range from an increase to a significant reduction in the nNOS activity in aged rat or mice models [136]. Mitochondrial investigations of NO effects support the hypothesis that NO levels could be increased during aging: endogenously-produced NO can inhibit mitochondrial respiration at cytochrome oxidase with the Ki ~50 nM (physiological range of NO concentrations is 0.1–100 nM) [137][138]. Moreover, NO can irreversibly suppress mitochondrial respiration, increase the production of ROS and RNS, and induce the mitochondrial permeability transition pore [137][138].
In the CNS, glutamate activates the N-methyl-d-aspartic acid (NMDA) receptor, which facilitates the influx of Ca2+ into the cell. The binding of Ca2+ to calmodulin activates the NOS production of NO which further activates the soluble guanylate cyclase and production of cGMP [139]. One of the main causes of dementia development is the aggregation of amyloid β (Aβ) peptides [140][141]. It has been shown that Aβ peptide interferes with the calmodulin-dependent NO synthesis. Chalimoniuk and Strosznajder (1998) showed that with advanced age, phosphodiesterase degradation of cGMP increased, which resulted in the decreased basal levels of cGMP. Moreover, the inhibition of cGMP-phosphodiesterase did not restore the NMDA-induced production of cGMP in the hippocampus and cerebellum of aged rats. However, the brain cortex in aged animals responded to the inhibition of cGMP-phosphodiesterase and showed a preserved production of cGMP. Interestingly, the NOS activity in the aged brain hippocampus and cerebellum was elevated by 175 and 160%, respectively. The treatment with Aβ fragment, peptide 25–35, reduced the Ca2+ transfer mediated by NMDA and thus calmodulin-dependent NO formation. The elevated NOS activity, despite disturbed cGMP-dependent signaling in the hippocampus and cerebellum of aged animals, might affect learning and memory function during aging, and the accumulation of Aβ during aging might be the cause of these disorders [142]. Activity-related Ca2+/calmodulin-dependent protein kinase II (CaMKII) autophosphorylation and AMPA receptor phosphorylation were reported as having crucial implications for hippocampal dentate long-term potentiation. Interruption in these mechanisms could directly associate with Aβ-induced impairments in memory and hippocampal synaptic plasticity [143][144].
Under chronic neuroinflammation conditions such as brain aging or neurodegenerative diseases, excessive levels of NO can be found at the choroid plexus (CP) via iNOS mediation, an epithelial layer that forms the blood-cerebrospinal fluid barrier (BCSFB). Glial cells are the source for these NO when they are chronically activated under the conditions such as aging or neurodegeneration [145][146][147]. Leukocytes reduce the neuroinflammation by entering CSF through the CP gateway; however, leukocytes do not always enter the CSF optimally during neuroinflammatory conditions. Thus, increased levels of leukocytes in the CSF can act as a potential therapeutic strategy [148][149][150][151][152][153][154]. It was suggested that NO alone in pathologic levels did not lead to neuronal death and gliosis. NO together with other cytotoxic and pro-inflammatory factors could induce neurodegeneration [147].
Baruch et al. (2015) showed that NO suppressed the leukocyte trafficking in choroid plexus (CP) by inhibiting epithelial nuclear factor-κB (NF-κB) p65 nuclear translocation in AD-Tg mice. Furthermore, the administration of the NO scavenger, rutin, enhanced the levels of leukocytes in the brain [155].
The tolerance of the brain towards hypoxemic conditions reduces by aging [156]. One of the suggested mechanisms for this phenomenon was the increase of free radicals during hypoxia and even at the reoxygenation period [157]. Hypoxic/ischemic conditions, including stroke, aneurysm, trauma, and infection, are among the most common causes of CNS dysfunction [158]. The increased frequency of hypoxic conditions during aging, along with the dysfunctional activity of the aged brain during these situations, highlight the importance of understanding the underlying mechanism behind these malfunctions. It has been shown that, contrary to eNOS and nNOS, which are constitutively expressed, iNOS induction is enhanced under conditions such as hypoxia [159][160]. Molina et al. (2017) studied the onset of the changes in the gene expression of NOS isoforms and their protein levels, as well as NO level changes following striatum hypoxia/reoxygenation in aged rats. Rats were exposed to hypoxemic conditions for 20 min and then were exposed to reoxygenation for 0 h, 24 h, and 5 days, in which they were sacrificed immediately thereafter. NOS gene expression levels did not change at 0 h following hypoxia. After 24 h, eNOS gene expression and protein levels, along with nNOS protein levels, were increased in the aged rat striatum. On day 5 of the post-hypoxia, iNOS gene expression did not change significantly, while iNOS protein levels were increased. Moreover, eNOS gene expression was enhanced while nNOS gene expression was suppressed. The levels of NO did not change significantly in aged striatum after hypoxia and reoxygenation, despite the increased activities of NOS isoforms during this time span. Molina et al. (2017) suggested that aging might disrupt the production of NO by NOS via decreasing the availability of BH4 cofactor and the impairment of Ca2+ homeostasis. Moreover, reduced O2 levels following hypoxia amplified the decrease in the NO production by NOS. Furthermore, the decreased NO in the hypoxic striatum could disturb the NO vasodilatory response in the brain vessels, which could further increase the severity of the hypoxic damage to the brain. The increased activity of NOS following hypoxia in the aged striatum was thought to be the compensatory mechanism of the brain to combat the effects of hypoxia on reducing O2 levels in the brain. Due to the impaired production of NO by NOS in the aged brain, the O2-independent production of NO was increased by denitrating the nitrated tyrosine residues to produce NO. The O2-independent production of NO in the aged striatum was shown as a continuous decline in the nitrated proteins from 0 h until 5 days after hypoxia/reoxygenation [161]. Aging-related changes in gene expression and corticostriatal synaptic plasticity were examined by Chepkova et al. (2015) in the dorsal striatum of four mice groups, aged from young to old [162]. The findings revealed a substantial drop in transcripts encoding neuronal NOS and activation receptors (NR1 glutamate NMDA receptor subunit and D1 dopamine receptor) in aged mice (Figure 4).
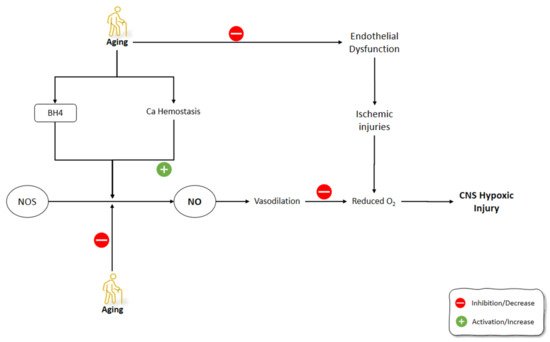
Figure 4. Effects of aging on NO production mechanisms and signaling pathways involved in the development of CNS ischemic injuries. NO: Nitric Oxide; NOS: Nitric Oxide Synthase; BH4: (6r-)-tetrahydro-l-biopterin.
3.3. Reproduction System Aging and NO
Estrogen can upregulate the endothelium-derived vasodilator factors, including NO and prostacyclin [163][164]. Following the estrogen drop after menopause, women might have accelerated vascular dysfunction, which means that estrogen can be a trigger for these processes [165][166]. The overlap of menopause with aging brought up the debate of whether the increased risk of cardiovascular disease in this period results from aging or menopause or both [167].
A vasoconstrictor prostanoid, thromboxane A2 (TXA2), activates the TXA2 receptor (TP) and has a primary role in the hemostasis of normal systemic vasculature [168][169]. Cyclooxygenase (COX) activation and elevated TP receptor expression are implicated in age-associated vascular diseases [170]. In humans, the production of COX-derived, endothelium-derived contractile factors is a feature of the aged blood vessels resulting in an earlier onset and speeding up of endothelial dysfunction [171].
NO can suppress the prostanoid production through the regulation of the COX activity and, conversely, the prostanoids, such as TXA2, can decrease the NOS expression and activity [172][173]. During the aging processes, endothelium-derived TXA2 increases vasoconstriction and endothelial dysfunction, which is amplified by decreased NO activity [174].
Vidal-Gómez et al. (2016) showed that in the absence of estrogen in young and aged ovariectomized mice, TP activation decreased NO bioavailability in the aorta by stimulating COX and the production of superoxide. The administration of estradiol restored the decreased NO bioavailability in the aorta due to TP activation. COX upregulation contributed partially to TXA2 contractile functions, which could be enhanced by aging and the absence of estrogen, while it could be suppressed due to the inhibition of COX by indomethacin. The production of superoxide, and therefore decreased NO bioavailability by endothelium-derived prostanoid TXA2 and TP activation, was also age- and estrogen-dependent, and could be prevented by suppressing COX. Interestingly, when estrogen was absent, COX inhibition resulted in enhanced bioavailability of NO [175].
Novensà et al. (2011) studied how aging alters the estrogen affects the NO development in an accelerated senescence mouse model [176]. Aged animals were similar to young animals in terms of DAF-2 or plasma nitrite/nitrate (NO2/NO3) in NO production. Estrogen treatment improved the production of NO in young animals by increasing eNOS expression but did not affect NO production in aged animals. Estrogen inhibited NADPH-oxidase and its production of superoxide anion (O2−). O2− catabolizes the NO and decreases its levels. Therefore, estrogen increased NO levels by decreasing NO catabolism via O2− through inhibiting the production of O2− by NADPH-oxidase. In Novensà et al.’s (2011) study, estrogen reduced the development of O2− in young female animals, while in the aged animals estrogen increased O2− [176]. The latter was due to the upregulated estrogen receptor (ERb/ERa) expression ratios in the aged female animals and thus modulated the antioxidant effects of estrogen, enhancing its pro-oxidant activity. The capacity of E2 to cause the modulation of eNOS and a decline of O2− was impaired by aging. Activation of ERα has been associated with augmented eNOS expression and NO formation, and its antioxidant mechanism of action. The recent studies on the ERα knockout mice demonstrated its importance in the regulation of eNOS. In the opposite, the knockout of ERβ caused the development of hypertension, regardless of its normal NO production. These outcomes suggested that ERα is the most important ER subtype accountable for NO formation by estrogen. Furthermore, augmented levels of ERβ could be related to cardiovascular risk, comprising coronary calcification and atherosclerosis [176].
Banerjee et al. (2012) showed that aromatase and ER-alpha were localized in Leydig cells of the testis and demonstrated a strong association between testicular aromatase and the level of circulating testosterone, implying that testicular steroidogenesis could be modulated by estrogen. The baseline level of NO during the reproductive activity cycle was shown in this study, although aged animals showed the correlation between decreased testicular activity and elevated serum NO. This research found that aromatase and NO levels were inversely associated. Furthermore, NO elevation downregulated steroidogenesis and germ cell survival. In summary, reduced estrogen enhances NO production in old age, which reduces testicular steroidogenesis and the apoptosis of germ cells [177] (Figure 5).
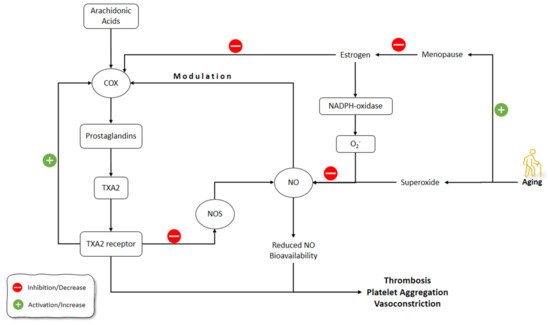
Figure 5. Effects of aging on estrogen and NO pathways involved in vasculature functions. NO: Nitric Oxide; NOS: Nitric Oxide Synthase; COX: Cyclooxygenase; TXA2: thromboxane A2.
One of the causes of reproductive failure is the oocyte’s postovulatory aging [178][179]. NO is a critical intracellular and intercellular messenger adjusting numerous physiological components in the processes of folliculogenesis, ovulation, and oviductal travel [180][181][182][183]. Goud et al. (2005) showed that NO exposure to young and old mice oocytes slowed the aging processes and increased the stability of microtubular spindle apparatus. The underlying mechanisms for these effects of NO were the activation of guanylate cyclase, which led to increased cGMP production [184]. Previous studies have shown that GMP alone can alleviate meiosis in rat and hamster oocytes. According to the outcomes of the aforementioned research, the production of cGMP occurred in the cumulus cells and was possibly transported into the oocyte via gap junctions to impede meiosis [185][186]. Similarly, specific PDEs are also triggered by cGMP and contribute to cAMP decrease [187]. This might activate the M-phase-promoting factor (MPF), a core regulator of M II phase arrest [188][189]. During the fertilization of oocytes, cortical granule depletion has been associated with aging-like changes such as elevated cytosolic Ca2+ and activated protein kinase C; however, these changes can occur in unfertilized oocytes due to aging [190][191]. In Goud et al.’s (2005) study, the mechanism of anti-aging effects of NO in oocytes might be related to the increase in Ca2+ levels or the suppression of protein kinase C activation [184] (Figure 6).
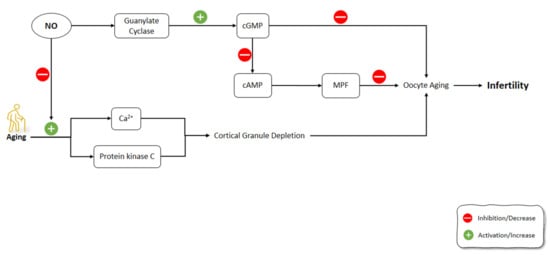
Figure 6. Effects of aging on the signaling pathways involved in oocytes aging including NO pathway. NO: Nitric Oxide; NOS: Nitric Oxide Synthase; cGMP: Cyclic guanosine monophosphate; cAMP: Cyclic adenosine monophosphate; MPF: M-phase-promoting factor.
3.4. Skin Aging and NO
In aged people, skin loses its ability to increase blood flow during heat stress, which can lead to the increased occurrence of heat-related diseases and even to death [192][193][194][195][196]. The sympathetic nervous system regulates the blood flow to the skin via the adrenergic vasoconstrictor system and an active vasodilator system [197].
An increase in body core temperature releases tonic adrenergic vasoconstrictor which elevates the skin blood flow. After a threshold, as temperature rises, sweating and cutaneous active vasodilation (AVD) occur [197]. In young individuals, about 30% of AVD is controlled by NO [198]. In older populations, NO-dependent vasodilation of skin is compromised [199]. Holowatz et al. (2003) showed that during passive whole-body heating, NO-dependent AVD was 23% and 60% in young and old individuals, respectively. Thus, the NO-mediated pathways were related more to the total vasodilatory response of the aged subjects at high core temperatures. The authors suggested that an unknown transmitter(s) could mediate the AVD during passive whole-body heating in aged individuals [200].
In another study, Bruning et al. (2012) showed that eNOS is primarily responsible for mediating cutaneous NO-dependent vasodilation during the local heating and perfusion of the endothelium-dependent agonist ACh in middle-aged skin. In addition, the expression of NOS isoforms or downstream vasodilatory molecular targets did not differ in the biopsy studies [201][202].
3.5. Renal Aging and NO
Reckelhoff et al. (1994) showed that, during aging, NO production decreases, which could be explained by the lack of the endogenous substrate in aged rats, l-arginine [203].
Age-dependent kidney diseases are more common in male humans. NO signaling dysfunction can cause age-dependent kidney damage. Erdely et al. (2003) found that renal NOS decreased by aging in male but not female rats [204]. These data suggested that the difference of NOS activity and NO function might be a determining factor for the gender gap in the rate of renal functional decline in advancing age. According to the study, a noticeable sexual dimorphism demonstrated that the females were protected both by the presence of estrogens and the deficiency of harmful androgens. The total NO formation, the activity of NOS and its abundance were important in the aging male rat kidney. Although arginine production was conserved, which showed elevation in the circulating NOS inhibitor, ADMA, in the aging male rat, these effects improved deteriorations in NO production [204][205].
Doleželová et al. (2016) showed that renal NO and CYP450-epoxygenase products, which counteract the development of hypertension and protect the kidney, were decreased in a genetic animal model of spontaneous hypertension (fawn-hooded hypertensive or FHH rat). Renal NOS activity was higher in adult animals with established hypertension but was similar between prehypertensive FHH rats and age-matched non-hypertensive controls [206]. Reduced vasodilator response in isolated renal vessels from adult rats has been shown in in vitro studies [207][208].
Stadler et al. (2003) demonstrated that before the occurrence of histopathological injuries in diabetes, NO and peroxynitrite are increased in streptozotocin-induced diabetic rats. Moreover, the ability of increased NO levels to suppress the formation of ROS in vessels and kidney tissues highlighted the possible roles of NO and peroxynitrite in late diabetic complications [92].
Impaired angiogenesis is believed to cause the age-related nephropathy, along with the gradual weakening of renal microvasculature. Satoh et al. (2013) showed that human umbilical vein endothelial cells (HUVECs) treated with NOS inhibitor had higher expression and activity of cathepsin D. Cathepsin D produces angiostatin, a potent in vivo angiogenesis inhibitor. Aged rats treated with l-NAME, had higher cathepsin D and angiostatin activity. On the other hand, treatment with a long-lasting NO-releasing vasodilator diminished the production and activity of cathepsin D and angiostatin. It was suggested that one of the most important steps to generate angiostatin would be related to the contribution of the proteolytic cleavage of plasminogen. Besides, the inhibition of cyclooxygenase (COX) and MMP-2 activities were also related to the angiostatin-generating mechanisms in vivo. In summary, cathepsin D-induced production of angiotensin was increased in the aging rat kidney. Decreased NO output triggered activity of cathepsin D. Increased development of angiostatin in the aged rat kidney could be due to the capillary loss and interstitial damage. [209]. However, in the aforementioned study, no direct interplay was observed between the generation of angiostatin in tubulointerstitial damage related to the peritubular capillary loss in the aged kidney.
3.6. Thyroid Aging and NO
The impairment of water reabsorption and improvements of aquaporin type 2 water channel (AQP2) were consistent with hypothyroid state and aging. AQP2 trafficking to the apical plasma membrane, in medullary duct collection cells, involves NO. Sarati et al. (2013) showed that the urine output and medullary NOS activity are correlated reversely in young and old rats, as the former was increased in the young and later was increased in the old rats. Elevated AQP2 in rats with hyperthyroidism was found in the plasma membrane of the young rats and at the cytosolic site in adult rats. These findings showed that medullary NO and AQP2 are implicated in maintaining water homeostasis [210]. Due to the role of the connection of NO-AQP2-hypothyroid damage in the regulation of water homeostasis, it was suggested that hypothyroidism may affect renal parameters in the aging processes.
3.7. Erectile Dysfunction, Aging, and NO
The decrease of smooth muscle cells (SMCs) and collagen in the corpus cavernosum (CC) greatly contributes to erectile dysfunction (ED) during aging. One of the occurring phenomena during the aging process is tissue remodeling in the CC. The aforementioned tissue remodeling feasibly occurs by reason of phenotypic cellular alteration from SMCs to fibroblasts that latterly would cause an elevation in the content of collagen deposit [211]. According to anatomical localization, the corpora cavernosa are paired spongy cylinders that lay on the superior facet of the penis [212]. The accumulation of collagen is thought to be affected by ROS. The NO production by iNOS has been shown to suppress the ROS and thus decrease collagen accumulation [211]. To clearly describe the relationship between ROS formation and collagen accumulation, this process was described in Peyronie’s disease, which is accompanied by fibrosis and is characterized by an augmentation in collagen over the intracellular part. Fibrosis is related to the formation of profibrotic factors, including plasminogen activator inhibitor-1, transforming growth factor beta (TGF-β), and ROS throughout oxidative stress. This is accompanied by the stimulation of the iNOS, which plays a crucial part as an endogenous antifibrotic pathway in the case of exposure with profibrotic manners [213]. Inside the arterial media SMC, Ferrini et al. (2004) explored the possibility of related mechanisms that exist within aging and that share specific common physiological roles with the cavernosal SMC [214]. Male, brown Norwegian rats aged (22–24 months) were treated with an iNOS action inhibitor. Inhibition of iNOS activity was induced by l-N-(iminoethyl)-lysine acetate. Resistance arteries of the penis showed an increased SMC apoptosis, increased collagen amounts, and elevated ROS and iNOS levels. Administration of an iNOS action inhibitor worsened the SMC/collagen ratio and increased ROS levels. A prevalent etiology can be explained in the hypotheses of ED and arteriosclerosis in aging males, namely that the SMC’s production of iNOS is an effort to combat this fibrosis [214].
This entry is adapted from the peer-reviewed paper 10.3390/molecules26154533
References
- Forstermann, U.; Sessa, W. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2011, 33, 829–837.
- Mikkelsen, R.B.; Wardman, P. Biological chemistry of reactive oxygen and nitrogen and radiation-induced signal transduction mechanisms. Oncogene 2003, 22, 5734–5754.
- Lee, J.H.; Yang, E.S.; Park, J.W. Inactivation of NADP+-dependent isocitrate dehydrogenase by peroxynitrite. Implications for cytotoxicity and alcohol-induced liver injury. J. Biol. Chem. 2003, 278, 51360–51371.
- Talebi, M.; Zarshenas, M.; Yazdani, E.; Moein, M. Preparation and evaluation of possible antioxidant activities of Rose traditional tablet “[Qurs-e-Vard]” a selected Traditional Persian Medicine [TPM] formulation via various procedures. Curr. Drug Discov. Technol. 2020, 17, 1–8.
- Talebi, M.; Kakouri, E.; Talebi, M.; Tarantilis, P.A.; Farkhondeh, T.; İlgün, S.; Pourbagher-Shahri, A.M.; Samargahndian, S. Nutraceuticals-based therapeutic approach: Recent advances to combat pathogenesis of Alzheimer’s disease. Expert Rev. Neurother. 2021, 21.
- Ahmad, R.; Hussain, A.; Ahsan, H. Peroxynitrite: Cellular pathology and implications in autoimmunity. J. Immunoass. Immunochem. 2019, 40, 1–16.
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424.
- Kar, S.; Kavdia, M. Endothelial NO and O2− production rates differentially regulate oxidative, nitroxidative, and nitrosative stress in the microcirculation. Free Radic. Biol. Med. 2013, 63, 161–174.
- Islam, B.U.; Habib, S.; Ahmad, P.; Allarakha, S.; Moinuddin; Ali, A. Pathophysiological Role of Peroxynitrite Induced DNA Damage in Human Diseases: A Special Focus on Poly(ADP-ribose) Polymerase (PARP). Indian J. Clin. Biochem. 2015, 30, 368–385.
- Shnayder, N.A.; Petrova, M.M.; Popova, T.E.; Davidova, T.K.; Bobrova, O.P.; Trefilova, V.V.; Goncharova, P.S.; Balberova, O.V.; Petrov, K.V.; Gavrilyuk, O.A.; et al. Prospects for the Personalized Multimodal Therapy Approach to Pain Management via Action on NO and NOS. Molecules 2021, 26, 2431.
- Farkhondeh, T.; Llorens, S.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Talebi, M.; Shakibaei, M.; Samarghandian, S. An Overview of the Role of Adipokines in Cardiometabolic Diseases. Molecules 2020, 25, 5218.
- Maccallini, C.; Mollica, A.; Amoroso, R. The Positive Regulation of eNOS Signaling by PPAR Agonists in Cardiovascular Diseases. Am. J. Cardiovasc. Drugs 2017, 17, 273–281.
- Ledo, A.; Lourenço, C.F.; Cadenas, E.; Barbosa, R.M.; Laranjinha, J. The bioactivity of neuronal-derived nitric oxide in aging and neurodegeneration: Switching signaling to degeneration. Free Radic. Biol. Med. 2021, 162, 500–513.
- Noble, M.A.; Munro, A.W.; Rivers, S.L.; Robledo, L.; Daff, S.N.; Yellowlees, L.J.; Shimizu, T.; Sagami, I.; Guillemette, J.G.; Chapman, S.K. Potentiometric analysis of the flavin cofactors of neuronal nitric oxide synthase. Biochemistry 1999, 38, 16413–16418.
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536.
- Crane, B.R.; Arvai, A.S.; Ghosh, D.K.; Wu, C.; Getzoff, E.D.; Stuehr, D.J.; Tainer, J.A. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science 1998, 279, 2121–2126.
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615.
- Cho, H.J.; Xie, Q.W.; Calaycay, J.; Mumford, R.A.; Swiderek, K.M.; Lee, T.D.; Nathan, C. Calmodulin is a subunit of nitric oxide synthase from macrophages. J. Exp. Med. 1992, 176, 599–604.
- Hemmens, B.; Mayer, B. Enzymology of nitric oxide synthases. Methods Mol. Biol. 1998, 100, 1–32.
- Zoupa, E.; Pitsikas, N. The Nitric Oxide (NO) Donor Sodium Nitroprusside (SNP) and Its Potential for the Schizophrenia Therapy: Lights and Shadows. Molecules 2021, 26, 3196.
- Rybalkin, S.D.; Yan, C.; Bornfeldt, K.E.; Beavo, J.A. Cyclic GMP Phosphodiesterases and Regulation of Smooth Muscle Function. Circ. Res. 2003, 93, 280–291.
- Zhou, L.; Zhu, D.-Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide 2009, 20, 223–230.
- Costa, E.D.; Rezende, B.A.; Cortes, S.F.; Lemos, V.S. Neuronal Nitric Oxide Synthase in Vascular Physiology and Diseases. Front. Physiol. 2016, 7.
- O’Toole, E.; Doucet, M.V.; Sherwin, E.; Harkin, A. Chapter 3—Novel Targets in the Glutamate and Nitric Oxide Neurotransmitter Systems for the Treatment of Depression. In Systems Neuroscience in Depression; Frodl, T., Ed.; Academic Press: San Diego, CA, USA, 2016; pp. 81–113.
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131.
- Förstermann, U. Regulation of nitric oxide synthase expression and activity. In Nitric Oxide: Biology and Chemistry; Springer: Berlin/Heidelberg, Germany, 2000; pp. 71–91.
- Hayashi, Y.; Nishio, M.; Naito, Y.; Yokokura, H.; Nimura, Y.; Hidaka, H.; Watanabe, Y. Regulation of Neuronal Nitric-oxide Synthase by Calmodulin Kinases. J. Biol. Chem. 1999, 274, 20597–20602.
- Kim, N.; Azadzoi, K.M.; Goldstein, I.; Saenz de Tejada, I. A nitric oxide-like factor mediates nonadrenergic-noncholinergic neurogenic relaxation of penile corpus cavernosum smooth muscle. J. Clin. Investig. 1991, 88, 112–118.
- Rajfer, J.; Aronson, W.J.; Bush, P.A.; Dorey, F.J.; Ignarro, L.J. Nitric oxide as a mediator of relaxation of the corpus cavernosum in response to nonadrenergic, noncholinergic neurotransmission. N. Engl. J. Med. 1992, 326, 90–94.
- Turko, I.V.; Ballard, S.A.; Francis, S.H.; Corbin, J.D. Inhibition of cyclic GMP-binding cyclic GMP-specific phosphodiesterase (Type 5) by sildenafil and related compounds. Mol. Pharmacol. 1999, 56, 124–130.
- Talebi, M.; Talebi, M.; Farkhondeh, T.; Simal-Gandara, J.; Kopustinskiene, D.M.; Bernatoniene, J.; Samarghandian, S. Emerging cellular and molecular mechanisms underlying anticancer indications of chrysin. Cancer Cell Int. 2021, 21, 214.
- Nathan, C.F.; Hibbs, J.B., Jr. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 1991, 3, 65–70.
- Talebi, M.; Talebi, M.; Farkhondeh, T.; Samarghandian, S. Molecular mechanism-based therapeutic properties of honey. Biomed. Pharmacother. 2020, 130, 110590.
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698.
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429.
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230.
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S.; et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991, 254, 1001–1003.
- Fehsel, K.; Jalowy, A.; Qi, S.; Burkart, V.; Hartmann, B.; Kolb, H. Islet cell DNA is a target of inflammatory attack by nitric oxide. Diabetes 1993, 42, 496–500.
- Li, L.M.; Kilbourn, R.G.; Adams, J.; Fidler, I.J. Role of nitric oxide in lysis of tumor cells by cytokine-activated endothelial cells. Cancer Res. 1991, 51, 2531–2535.
- Green, S.J.; Mellouk, S.; Hoffman, S.L.; Meltzer, M.S.; Nacy, C.A. Cellular mechanisms of nonspecific immunity to intracellular infection: Cytokine-induced synthesis of toxic nitrogen oxides from l-arginine by macrophages and hepatocytes. Immunol. Lett. 1990, 25, 15–19.
- Song, Y.; Cardounel, A.J.; Zweier, J.L.; Xia, Y. Inhibition of superoxide generation from neuronal nitric oxide synthase by heat shock protein 90: Implications in NOS regulation. Biochemistry 2002, 41, 10616–10622.
- Pritchard, K.A., Jr.; Ackerman, A.W.; Gross, E.R.; Stepp, D.W.; Shi, Y.; Fontana, J.T.; Baker, J.E.; Sessa, W.C. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J. Biol. Chem. 2001, 276, 17621–17624.
- Ratajczak, P.; Damy, T.; Heymes, C.; Oliviéro, P.; Marotte, F.; Robidel, E.; Sercombe, R.; Boczkowski, J.; Rappaport, L.; Samuel, J.L. Caveolin-1 and -3 dissociations from caveolae to cytosol in the heart during aging and after myocardial infarction in rat. Cardiovasc. Res. 2003, 57, 358–369.
- Sowa, G.; Pypaert, M.; Sessa, W.C. Distinction between signaling mechanisms in lipid rafts vs. caveolae. Proc. Natl. Acad. Sci. USA 2001, 98, 14072–14077.
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452.
- Gratton, J.P.; Fontana, J.; O’Connor, D.S.; Garcia-Cardena, G.; McCabe, T.J.; Sessa, W.C. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro. Evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J. Biol. Chem. 2000, 275, 22268–22272.
- Fulton, D.; Gratton, J.P.; McCabe, T.J.; Fontana, J.; Fujio, Y.; Walsh, K.; Franke, T.F.; Papapetropoulos, A.; Sessa, W.C. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 1999, 399, 597–601.
- Fleming, I.; Busse, R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1–R12.
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 2011, 589, 4545–4554.
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290–2307.
- Mattagajasingh, I.; Kim, C.-S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.-B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860.
- Montesanto, A.; Crocco, P.; Tallaro, F.; Pisani, F.; Mazzei, B.; Mari, V.; Corsonello, A.; Lattanzio, F.; Passarino, G.; Rose, G. Common polymorphisms in nitric oxide synthase (NOS) genes influence quality of aging and longevity in humans. Biogerontology 2013, 14, 177–186.
- Cooke, J.P.; Dzau, J.; Creager, A. Endothelial dysfunction in hypercholesterolemia is corrected by l-arginine. Basic Res. Cardiol. 1991, 86 (Suppl. S2), 173–181.
- Harrison, D.G. Cellular and molecular mechanisms of endothelial cell dysfunction. J. Clin. Investig. 1997, 100, 2153–2157.
- Wu, G.; Meininger, C.J. Arginine nutrition and cardiovascular function. J. Nutr. 2000, 130, 2626–2629.
- Pourbagher-Shahri, A.M.; Farkhondeh, T.; Ashrafizadeh, M.; Talebi, M.; Samargahndian, S. Curcumin and cardiovascular diseases: Focus on cellular targets and cascades. Biomed. Pharmacother. 2021, 136, 111214.
- Hecker, M.; Nematollahi, H.; Hey, C.; Busse, R.; Racké, K. Inhibition of arginase by NG-hydroxy-l-arginine in alveolar macrophages: Implications for the utilization of l-arginine for nitric oxide synthesis. FEBS Lett. 1995, 359, 251–254.
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 2003, 108, 2000–2006.
- Chou, T.C.; Yen, M.H.; Li, C.Y.; Ding, Y.A. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension 1998, 31, 643–648.
- Wu, C.C.; Yen, M.H. Higher level of plasma nitric oxide in spontaneously hypertensive rats. Am. J. Hypertens. 1999, 12, 476–482.
- Vaziri, N.D.; Ni, Z.; Oveisi, F. Upregulation of renal and vascular nitric oxide synthase in young spontaneously hypertensive rats. Hypertension 1998, 31, 1248–1254.
- Wu, C.-C.; Hong, H.-J.; Chou, T.-C.; Ding, Y.-A.; Yen, M.-H. Evidence for Inducible Nitric Oxide Synthase in Spontaneously Hypertensive Rats. Biochem. Biophys. Res. Commun. 1996, 228, 459–466.
- Cernadas, M.R.; Sánchez de Miguel, L.; García-Durán, M.; González-Fernández, F.; Millás, I.; Montón, M.; Rodrigo, J.; Rico, L.; Fernández, P.; de Frutos, T.; et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ. Res. 1998, 83, 279–286.
- Zieman, S.J.; Gerstenblith, G.; Lakatta, E.G.; Rosas, G.O.; Vandegaer, K.; Ricker, K.M.; Hare, J.M. Upregulation of the nitric oxide-cGMP pathway in aged myocardium: Physiological response to l-arginine. Circ. Res. 2001, 88, 97–102.
- Gerassimou, C.; Kotanidou, A.; Zhou, Z.; Simoes, D.C.M.; Roussos, C.; Papapetropoulos, A. Regulation of the expression of soluble guanylyl cyclase by reactive oxygen species. Br. J. Pharmacol. 2007, 150, 1084–1091.
- Pizzarelli, F.; Maas, R.; Dattolo, P.; Tripepi, G.; Michelassi, S.; D’Arrigo, G.; Mieth, M.; Bandinelli, S.; Ferrucci, L.; Zoccali, C. Asymmetric dimethylarginine predicts survival in the elderly. Age 2013, 35, 2465–2475.
- Sverdlov, A.L.; Ngo, D.T.M.; Chan, W.P.A.; Chirkov, Y.Y.; Horowitz, J.D. Aging of the nitric oxide system: Are we as old as our NO? J. Am. Heart Assoc. 2014, 3, e000973.
- Scalera, F.; Martens-Lobenhoffer, J.; Täger, M.; Bukowska, A.; Lendeckel, U.; Bode-Böger, S.M. Effect of l-arginine on asymmetric dimethylarginine (ADMA) or homocysteine-accelerated endothelial cell aging. Biochem. Biophys. Res. Commun. 2006, 345, 1075–1082.
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415.
- Thijssen, D.H.; Hopman, M.T.; Levine, B.D. Endothelin and aged blood vessels: One more reason to get off the couch? Hypertension 2007, 50, 292–293.
- Thijssen, D.H.; Rongen, G.A.; van Dijk, A.; Smits, P.; Hopman, M.T. Enhanced endothelin-1-mediated leg vascular tone in healthy older subjects. J. Appl. Physiol. 2007, 103, 852–857.
- Van Guilder, G.P.; Westby, C.M.; Greiner, J.J.; Stauffer, B.L.; DeSouza, C.A. Endothelin-1 vasoconstrictor tone increases with age in healthy men but can be reduced by regular aerobic exercise. Hypertension 2007, 50, 403–409.
- Tokunaga, O.; Fan, J.; Watanabe, T.; Kobayashi, M.; Kumazaki, T.; Mitsui, Y. Endothelin. Immunohistologic localization in aorta and biosynthesis by cultured human aortic endothelial cells. Lab. Investig. J. Tech. Methods Pathol. 1992, 67, 210–217.
- Donato, A.J.; Gano, L.B.; Eskurza, I.; Silver, A.E.; Gates, P.E.; Jablonski, K.; Seals, D.R. Vascular endothelial dysfunction with aging: Endothelin-1 and endothelial nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H425–H432.
- Muller-Delp, J.M.; Spier, S.A.; Ramsey, M.W.; Delp, M.D. Aging impairs endothelium-dependent vasodilation in rat skeletal muscle arterioles. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1662–H1672.
- Csiszar, A.; Ungvari, Z.; Edwards, J.G.; Kaminski, P.; Wolin, M.S.; Koller, A.; Kaley, G. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 2002, 90, 1159–1166.
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Salvetti, G.; Bernini, G.; Magagna, A.; Salvetti, A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension 2001, 38, 274–279.
- Hamilton, C.A.; Brosnan, M.J.; McIntyre, M.; Graham, D.; Dominiczak, A.F. Superoxide excess in hypertension and aging: A common cause of endothelial dysfunction. Hypertension 2001, 37, 529–534.
- Van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced peroxynitrite formation is associated with vascular aging. J. Exp. Med. 2000, 192, 1731–1744.
- Ungvari, Z.; Kaley, G.; de Cabo, R.; Sonntag, W.E.; Csiszar, A. Mechanisms of Vascular Aging: New Perspectives. J. Gerontol. Ser. A 2010, 65, 1028–1041.
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953.
- Didion, S.P.; Ryan, M.J.; Didion, L.A.; Fegan, P.E.; Sigmund, C.D.; Faraci, F.M. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ. Res. 2002, 91, 938–944.
- Sun, D.; Huang, A.; Yan, E.H.; Wu, Z.; Yan, C.; Kaminski, P.M.; Oury, T.D.; Wolin, M.S.; Kaley, G. Reduced release of nitric oxide to shear stress in mesenteric arteries of aged rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2249–H2256.
- Feron, O.; Balligand, J.-L. Caveolins and the regulation of endothelial nitric oxide synthase in the heart. Cardiovasc. Res. 2006, 69, 788–797.
- Feron, O.; Belhassen, L.; Kobzik, L.; Smith, T.W.; Kelly, R.A.; Michel, T. Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J. Biol. Chem. 1996, 271, 22810–22814.
- Arreche, N.D.; Sarati, L.I.; Martinez, C.R.; Fellet, A.L.; Balaszczuk, A.M. Contribution of caveolin-1 to ventricular nitric oxide in age-related adaptation to hypovolemic state. Regul. Pept. 2012, 179, 43–49.
- Barouch, L.A.; Harrison, R.W.; Skaf, M.W.; Rosas, G.O.; Cappola, T.P.; Kobeissi, Z.A.; Hobai, I.A.; Lemmon, C.A.; Burnett, A.L.; O’Rourke, B.; et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 2002, 416, 337–339.
- Arza, P.; Netti, V.; Perosi, F.; Cernadas, G.; Ochoa, F.; Magnani, N.; Evelson, P.; Zotta, E.; Fellet, A.; Balaszczuk, A.M. Involvement of nitric oxide and caveolins in the age-associated functional and structural changes in a heart under osmotic stress. Biomed. Pharmacother. 2015, 69, 380–387.
- Wei, J.Y. Age and the cardiovascular system. N. Engl. J. Med. 1992, 327, 1735–1739.
- Szoke, E.; Shrayyef, M.Z.; Messing, S.; Woerle, H.J.; van Haeften, T.W.; Meyer, C.; Mitrakou, A.; Pimenta, W.; Gerich, J.E. Effect of aging on glucose homeostasis: Accelerated deterioration of beta-cell function in individuals with impaired glucose tolerance. Diabetes Care 2008, 31, 539–543.
- Bouché, C.; Serdy, S.; Kahn, C.R.; Goldfine, A.B. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr. Rev. 2004, 25, 807–830.
- Stadler, K.; Jenei, V.; von Bölcsházy, G.; Somogyi, A.; Jakus, J. Increased nitric oxide levels as an early sign of premature aging in diabetes. Free Radic. Biol. Med. 2003, 35, 1240–1251.
- Ropelle, E.R.; Pauli, J.R.; Cintra, D.E.; da Silva, A.S.; De Souza, C.T.; Guadagnini, D.; Carvalho, B.M.; Caricilli, A.M.; Katashima, C.K.; Carvalho-Filho, M.A.; et al. Targeted Disruption of Inducible Nitric Oxide Synthase Protects Against Aging, S-Nitrosation, and Insulin Resistance in Muscle of Male Mice. Diabetes 2013, 62, 466.
- Herrera, M.D.; Mingorance, C.; Rodríguez-Rodríguez, R.; Alvarez de Sotomayor, M. Endothelial dysfunction and aging: An update. Ageing Res. Rev. 2010, 9, 142–152.
- Erusalimsky, J.D. Vascular endothelial senescence: From mechanisms to pathophysiology. J. Appl. Physiol. 2009, 106, 326–332.
- Hadi, H.A.; Suwaidi, J.A. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 2007, 3, 853–876.
- Deedwania, P.C. Mechanisms of endothelial dysfunction in the metabolic syndrome. Curr. Diabetes Rep. 2003, 3, 289–292.
- Rippe, C.; Blimline, M.; Magerko, K.A.; Lawson, B.R.; LaRocca, T.J.; Donato, A.J.; Seals, D.R. MicroRNA changes in human arterial endothelial cells with senescence: Relation to apoptosis, eNOS and inflammation. Exp. Gerontol. 2012, 47, 45–51.
- Tanaka, J.; Qiang, L.; Banks, A.S.; Welch, C.L.; Matsumoto, M.; Kitamura, T.; Ido-Kitamura, Y.; DePinho, R.A.; Accili, D. Foxo1 links hyperglycemia to LDL oxidation and endothelial nitric oxide synthase dysfunction in vascular endothelial cells. Diabetes 2009, 58, 2344–2354.
- Rogers, S.C.; Zhang, X.; Azhar, G.; Luo, S.; Wei, J.Y. Exposure to high or low glucose levels accelerates the appearance of markers of endothelial cell senescence and induces dysregulation of nitric oxide synthase. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 1469–1481.
- Yu, Q.; Gao, F.; Ma, X.L. Insulin says NO to cardiovascular disease. Cardiovasc. Res. 2011, 89, 516–524.
- Dimmeler, S.; Zeiher, A.M.J.C. Endothelial cell apoptosis in angiogenesis and vessel regression. Circ. Res. 2000, 87, 434–439.
- Dimmeler, S.; Haendeler, J.; Galle, J.; Zeiher, A.M.J.C. Oxidized low-density lipoprotein induces apoptosis of human endothelial cells by activation of CPP32-like proteases: A mechanistic clue to the ‘response to injury’hypothesis. Circulation 1997, 95, 1760–1763.
- Dimmeler, S.; Rippmann, V.; Weiland, U.; Haendeler, J.; Zeiher, A.M. Angiotensin II induces apoptosis of human endothelial cells: Protective effect of nitric oxide. Circ. Res. 1997, 81, 970–976.
- Dimmeler, S.; Haendeler, J.; Nehls, M.; Zeiher, A.M. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1β–converting enzyme (ice)-like and cysteine protease protein (cpp)-32–like proteases. J. Exp. Med. 1997, 185, 601–608.
- Tenneti, L.; D’Emilia, D.M.; Lipton, S.A. Suppression of neuronal apoptosis by S-nitrosylation of caspases. Neurosci. Lett. 1997, 236, 139–142.
- Li, J.; Billiar, T.R.; Talanian, R.V.; Kim, Y.M. Nitric oxide reversibly inhibits seven members of the caspase family via S-nitrosylation. Biochem. Biophys. Res. Commun. 1997, 240, 419–424.
- Hoffmann, J.; Haendeler, J.; Aicher, A.; Rössig, L.; Vasa, M.; Zeiher, A.M.; Dimmeler, S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: Important role of nitric oxide. Circ. Res. 2001, 89, 709–715.
- Oelze, M.; Kröller-Schön, S.; Steven, S.; Lubos, E.; Doppler, C.; Hausding, M.; Tobias, S.; Brochhausen, C.; Li, H.; Torzewski, M.; et al. Glutathione peroxidase-1 deficiency potentiates dysregulatory modifications of endothelial nitric oxide synthase and vascular dysfunction in aging. Hypertension 2014, 63, 390–396.
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997.
- Lei, X.G. In vivo antioxidant role of glutathione peroxidase: Evidence from knockout mice. In Methods in Enzymology; Sies, H., Packer, L., Eds.; Academic Press: Cambridge, MA, USA, 2002; Volume 347, pp. 213–225.
- Benigni, A.; Cassis, P.; Conti, S.; Perico, L.; Corna, D.; Cerullo, D.; Zentilin, L.; Zoja, C.; Perna, A.; Lionetti, V.; et al. Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxid. Redox Signal. 2019, 31, 1255–1271.
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48.
- Lu, H.; Sun, L.; Chen, W.; Zhou, Y.; Liu, K.; Chen, J.; Zhang, Z.; Zhang, C.; Tian, H. Sirtuin 3 Therapy Attenuates Aging Expression, Oxidative Stress Parameters, and Neointimal Hyperplasia Formation in Vein Grafts. Ann. Vasc. Surg. 2020, 64, 303–317.
- Owusu, B.Y.; Stapley, R.; Patel, R.P. Nitric oxide formation versus scavenging: The red blood cell balancing act. J. Physiol. 2012, 590, 4993–5000.
- May, J.M.; Qu, Z.C.; Xia, L.; Cobb, C.E. Nitrite uptake and metabolism and oxidant stress in human erythrocytes. Am. J. Physiol. Cell Physiol. 2000, 279, C1946–C1954.
- Piknova, B.; Keszler, A.; Hogg, N.; Schechter, A.N. The reaction of cell-free oxyhemoglobin with nitrite under physiologically relevant conditions: Implications for nitrite-based therapies. Nitric Oxide Biol. Chem. 2009, 20, 88–94.
- Subasinghe, W.; Spence, D.M. Simultaneous determination of cell aging and ATP release from erythrocytes and its implications in type 2 diabetes. Anal. Chim. Acta 2008, 618, 227–233.
- Owusu, B.Y.; Stapley, R.; Honavar, J.; Patel, R.P. Effects of erythrocyte aging on nitric oxide and nitrite metabolism. Antioxid. Redox Signal. 2013, 19, 1198–1208.
- Kedar, N.P. Can we prevent Parkinson’s and Alzheimer’s disease? J. Postgrad. Med. 2003, 49, 236.
- Peinado, M.A. Histology and histochemistry of the aging cerebral cortex: An overview. Microsc. Res. Tech. 1998, 43, 1–7.
- Knight, J.A. The process and theories of aging. Ann. Clin. Lab. Sci. 1995, 25, 1–12.
- Martínez, M.C.; Andriantsitohaina, R. Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease. Antioxid. Redox Signal. 2008, 11, 669–702.
- Talebi, M.; İlgün, S.; Ebrahimi, V.; Talebi, M.; Farkhondeh, T.; Ebrahimi, H.; Samarghandian, S. Zingiber officinale ameliorates Alzheimer’s disease and Cognitive Impairments: Lessons from preclinical studies. Biomed. Pharmacother. 2021, 133.
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407.
- Paakkari, I.; Lindsberg, P. Nitric oxide in the central nervous system. Ann. Med. 1995, 27, 369–377.
- Bredt, D.S.; Snyder, S.H. Transient nitric oxide synthase neurons in embryonic cerebral cortical plate, sensory ganglia, and olfactory epithelium. Neuron 1994, 13, 301–313.
- Zhuo, M.; Hawkins, R.D. Long-term depression: A learning-related type of synaptic plasticity in the mammalian central nervous system. Rev. Neurosci. 1995, 6, 259–277.
- Law, A.; Gauthier, S.; Quirion, R. Say NO to Alzheimer’s disease: The putative links between nitric oxide and dementia of the Alzheimer’s type. Brain Res. Rev. 2001, 35, 73–96.
- Uttenthal, L.O.; Alonso, D.; Fernández, A.P.; Campbell, R.O.; Moro, M.A.; Leza, J.C.; Lizasoain, I.; Esteban, F.J.; Barroso, J.B.; Valderrama, R.; et al. Neuronal and inducible nitric oxide synthase and nitrotyrosine immunoreactivities in the cerebral cortex of the aging rat. Microsc. Res. Tech. 1998, 43, 75–88.
- Hilbig, H.; Hüller, J.; Dinse, H.R.; Bidmon, H.-J. In contrast to neuronal NOS-I, the inducible NOS-II expression in aging brains is modified by enriched environmental conditions. Exp. Toxicol. Pathol. 2002, 53, 427–431.
- Talebi, M.; Talebi, M.; Kakouri, E.; Farkhondeh, T.; Pourbagher-Shahri, A.M.; Tarantilis, P.A.; Samarghandian, S. Tantalizing role of p53 molecular pathways and its coherent medications in neurodegenerative diseases. Int. J. Biol. Macromol. 2021, 172, 93–103.
- Lores-Arnaiz, S.; Bustamante, J. Age-related alterations in mitochondrial physiological parameters and nitric oxide production in synaptic and non-synaptic brain cortex mitochondria. Neuroscience 2011, 188, 117–124.
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431.
- Jung, J.; Na, C.; Huh, Y. Alterations in Nitric Oxide Synthase in the Aged CNS. Oxid. Med. Cell. Longev. 2012, 2012, 718976.
- Colas, D.; Gharib, A.; Bezin, L.; Morales, A.; Guidon, G.; Cespuglio, R.; Sarda, N. Regional age-related changes in neuronal nitric oxide synthase (nNOS), messenger RNA levels and activity in SAMP8 brain. BMC Neurosci. 2006, 7, 81.
- Brown, G.C.; Bal-Price, A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol. 2003, 27, 325–355.
- Brown, G.C.; Borutaite, V. Nitric oxide inhibition of mitochondrial respiration and its role in cell death. Free Radic. Biol. Med. 2002, 33, 1440–1450.
- Corbalán, R.; Llansola, M.; Monfort, P.; Montoliu, C.; Muñoz, M.-D.; Hernández-Viadel, M.; Erceg, S.; Sánchez-Pérez, A.; Felipo, V. Hyperammonemia and liver failure alter signal transduction associated with glutamate receptors and modulation of guanylate cyclase by nitric oxide. In Encephalopathy and Nitrogen Metabolism in Liver Failure; Springer: Dordrecht, The Netherlands, 2003; pp. 193–208.
- Bharadwaj, P.R.; Dubey, A.K.; Masters, C.L.; Martins, R.N.; Macreadie, I.G. Abeta aggregation and possible implications in Alzheimer’s disease pathogenesis. J. Cell. Mol. Med. 2009, 13, 412–421.
- Talebi, M.; Talebi, M.; Farkhondeh, T.; Mishra, G.; İlgün, S.; Samarghandian, S. New insights into the role of the Nrf2 signaling pathway in green tea catechin applications. Phytother. Res. 2021, 35, 3078–3112.
- Chalimoniuk, M.; Strosznajder, J.B. Aging modulates nitric oxide synthesis and cGMP levels in hippocampus and cerebellum. Effects of amyloid beta peptide. Mol. Chem. Neuropathol. 1998, 35, 77–95.
- Zhao, D.; Watson, J.B.; Xie, C.W. Amyloid beta prevents activation of calcium/calmodulin-dependent protein kinase II and AMPA receptor phosphorylation during hippocampal long-term potentiation. J. Neurophysiol. 2004, 92, 2853–2858.
- Ghosh, A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78.
- Bal-Price, A.; Matthias, A.; Brown, G.C. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J. Neurochem. 2002, 80, 73–80.
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69.
- Liu, B.; Gao, H.M.; Wang, J.Y.; Jeohn, G.H.; Cooper, C.L.; Hong, J.S. Role of nitric oxide in inflammation-mediated neurodegeneration. Ann. N. Y. Acad. Sci. 2002, 962, 318–331.
- Simard, A.R.; Soulet, D.; Gowing, G.; Julien, J.P.; Rivest, S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 2006, 49, 489–502.
- Mildner, A.; Schlevogt, B.; Kierdorf, K.; Böttcher, C.; Erny, D.; Kummer, M.P.; Quinn, M.; Brück, W.; Bechmann, I.; Heneka, M.T.; et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 11159–11171.
- Vaknin, I.; Kunis, G.; Miller, O.; Butovsky, O.; Bukshpan, S.; Beers, D.R.; Henkel, J.S.; Yoles, E.; Appel, S.H.; Schwartz, M. Excess circulating alternatively activated myeloid (M2) cells accelerate ALS progression while inhibiting experimental autoimmune encephalomyelitis. PLoS ONE 2011, 6, e26921.
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312.
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235.
- Popovich, P.G.; Longbrake, E.E. Can the immune system be harnessed to repair the CNS? Nat. Rev. Neurosci. 2008, 9, 481–493.
- Schwartz, M.; Baruch, K. Breaking peripheral immune tolerance to CNS antigens in neurodegenerative diseases: Boosting autoimmunity to fight-off chronic neuroinflammation. J. Autoimmun. 2014, 54, 8–14.
- Baruch, K.; Kertser, A.; Porat, Z.; Schwartz, M. Cerebral nitric oxide represses choroid plexus NFκB-dependent gateway activity for leukocyte trafficking. EMBO J. 2015, 34, 1816–1828.
- Mariani, J.; Ou, R.; Bailey, M.; Rowland, M.; Nagley, P.; Rosenfeldt, F.; Pepe, S. Tolerance to ischemia and hypoxia is reduced in aged human myocardium. J. Thorac. Cardiovasc. Surg. 2000, 120, 660–667.
- Rouschop, K.M.A.; Ramaekers, C.H.M.A.; Schaaf, M.B.E.; Keulers, T.G.H.; Savelkouls, K.G.M.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Autophagy is required during cycling hypoxia to lower production of reactive oxygen species. Radiother. Oncol. 2009, 92, 411–416.
- Acker, T.; Acker, H. Cellular oxygen sensing need in CNS function: Physiological and pathological implications. J. Exp. Biol. 2004, 207, 3171–3188.
- Rus, A.; Peinado, M.Á.; Castro, L.; Del Moral, M.L. Lung eNOS and iNOS are reoxygenation time-dependent upregulated after acute hypoxia. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2010, 293, 1089–1098.
- Rus, A.; Molina, F.; Peinado, M.Á.; Del Moral, M.L. Nitric oxide averts hypoxia-induced damage during reoxygenation in rat heart. Microsc. Res. Tech. 2011, 74, 1093–1103.
- Molina, F.; Del Moral, M.L.; Peinado, M.; Rus, A. Response of the Nitric Oxide System to Hypobaric Hypoxia in the Aged Striatum. Gerontology 2017, 63, 36–44.
- Chepkova, A.N.; Schönfeld, S.; Sergeeva, O.A. Age-related alterations in the expression of genes and synaptic plasticity associated with nitric oxide signaling in the mouse dorsal striatum. Neural Plast. 2015, 2015, 458123.
- Mendelsohn, M.E. Estrogen actions in the cardiovascular system. Climacteric 2009, 12, 18–21.
- Sobrino, A.; Oviedo, P.J.; Novella, S.; Laguna-Fernandez, A.; Bueno, C.; García-Pérez, M.A.; Tarín, J.J.; Cano, A.; Hermenegildo, C. Estradiol selectively stimulates endothelial prostacyclin production through estrogen receptor-α. J. Mol. Endocrinol. 2010, 44, 237.
- Moreau, K.L.; Stauffer, B.L.; Kohrt, W.M.; Seals, D.R. Essential Role of Estrogen for Improvements in Vascular Endothelial Function With Endurance Exercise in Postmenopausal Women. J. Clin. Endocrinol. Metab. 2013, 98, 4507–4515.
- Novella, S.; Dantas, A.P.; Segarra, G.; Vidal-Gómez, X.; Mompeón, A.; Garabito, M.; Hermenegildo, C.; Medina, P. Aging-related endothelial dysfunction in the aorta from female senescence-accelerated mice is associated with decreased nitric oxide synthase expression. Exp. Gerontol. 2013, 48, 1329–1337.
- Bittner, V. Postmenopausal hormone therapy and the risk of cardiovascular disease. Expert Opin. Pharmacother. 2009, 10, 2041–2053.
- Fulton, C.T.; Stallone, J.N. Sexual dimorphism in prostanoid-potentiated vascular contraction: Roles of endothelium and ovarian steroids. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2062–H2073.
- Li, M.; Kuo, L.; Stallone, J.N. Estrogen potentiates constrictor prostanoid function in female rat aorta by upregulation of cyclooxygenase-2 and thromboxane pathway expression. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2444–H2455.
- Novella, S.; Dantas, A.P.; Segarra, G.; Novensa, L.; Heras, M.; Hermenegildo, C.; Medina, P. Aging enhances contraction to thromboxane A2 in aorta from female senescence-accelerated mice. Age 2013, 35, 117–128.
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Magagna, A.; Salvetti, A. Cyclooxygenase Inhibition Restores Nitric Oxide Activity in Essential Hypertension. Hypertension 1997, 29, 274–279.
- Miyamoto, A.; Hashiguchi, Y.; Obi, T.; Ishiguro, S.; Nishio, A. Ibuprofen or ozagrel increases NO release and l-nitro arginine induces TXA2 release from cultured porcine basilar arterial endothelial cells. Vasc. Pharmacol. 2007, 46, 85–90.
- Ashton, A.W.; Ware, J.A. Thromboxane A2 receptor signaling inhibits vascular endothelial growth factor-induced endothelial cell differentiation and migration. Circ. Res. 2004, 95, 372–379.
- Félétou, M.; Verbeuren, T.J.; Vanhoutte, P.M. Endothelium-dependent contractions in SHR: A tale of prostanoid TP and IP receptors. Br. J. Pharmacol. 2009, 156, 563–574.
- Vidal-Gómez, X.; Novella, S.; Pérez-Monzó, I.; Garabito, M.; Dantas, A.P.; Segarra, G.; Hermenegildo, C.; Medina, P. Decreased bioavailability of nitric oxide in aorta from ovariectomized senescent mice. Role of cyclooxygenase. Exp. Gerontol. 2016, 76, 1–8.
- Novensà, L.; Novella, S.; Medina, P.; Segarra, G.; Castillo, N.; Heras, M.; Hermenegildo, C.; Dantas, A.P. Aging negatively affects estrogens-mediated effects on nitric oxide bioavailability by shifting ERα/ERβ balance in female mice. PLoS ONE 2011, 6, e25335.
- Banerjee, A.; Anjum, S.; Verma, R.; Krishna, A. Alteration in expression of estrogen receptor isoforms alpha and beta, and aromatase in the testis and its relation with changes in nitric oxide during aging in mice. Steroids 2012, 77, 609–620.
- Ducibella, T. The cortical reaction and development of activation competence in mammalian oocytes. Hum. Reprod. Update 1996, 2, 29–42.
- Goud, P.; Goud, A.; Van Oostveldt, P.; Van der Elst, J.; Dhont, M. Fertilization abnormalities and pronucleus size asynchrony after intracytoplasmic sperm injection are related to oocyte postmaturity. Fertil. Steril. 1999, 72, 245–252.
- Yamauchi, J.; Miyazaki, T.; Iwasaki, S.; Kishi, I.; Kuroshima, M.; Tei, C.; Yoshimura, Y. Effects of nitric oxide on ovulation and ovarian steroidogenesis and prostaglandin production in the rabbit. Endocrinology 1997, 138, 3630–3637.
- Jablonka-Shariff, A.; Olson, L.M. The role of nitric oxide in oocyte meiotic maturation and ovulation: Meiotic abnormalities of endothelial nitric oxide synthase knock-out mouse oocytes. Endocrinology 1998, 139, 2944–2954.
- Sengoku, K.; Takuma, N.; Horikawa, M.; Tsuchiya, K.; Komori, H.; Sharifa, D.; Tamate, K.; Ishikawa, M. Requirement of nitric oxide for murine oocyte maturation, embryo development, and trophoblast outgrowth in vitro. Mol. Reprod. Dev. 2001, 58, 262–268.
- Chun, S.Y.; Eisenhauer, K.M.; Minami, S.; Billig, H.; Perlas, E.; Hsueh, A.J. Hormonal regulation of apoptosis in early antral follicles: Follicle-stimulating hormone as a major survival factor. Endocrinology 1996, 137, 1447–1456.
- Goud, A.P.; Goud, P.T.; Diamond, M.P.; Abu-Soud, H.M. Nitric oxide delays oocyte aging. Biochemistry 2005, 44, 11361–11368.
- Törnell, J.; Billig, H.; Hillensjö, T. Regulation of oocyte maturation by changes in ovarian levels of cyclic nucleotides. Hum. Reprod. 1991, 6, 411–422.
- Hubbard, C.J.; Price, J. The effects of follicle-stimulating hormone and cyclic guanosine 3′,5′-monophosphate on cyclic adenosine 3′,5′-monophosphate-phosphodiesterase and resumption of meiosis in hamster cumulus-oocyte complexes. Biol. Reprod. 1988, 39, 829–838.
- Murad, F. Regulation of cytosolic guanylyl cyclase by nitric oxide: The NO-cyclic GMP signal transduction system. Adv. Pharmacol. 1994, 26, 19–33.
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol. Rev. 2000, 52, 375–414.
- Gotoh, Y.; Nishida, E. The MAP kinase cascade: Its role in Xenopus oocytes, eggs and embryos. Prog. Cell Cycle Res. 1995, 1, 287–297.
- Raz, T.; Skutelsky, E.; Amihai, D.; Hammel, I.; Shalgi, R. Mechanisms leading to cortical reaction in the mammalian egg. Mol. Reprod. Dev. 1998, 51, 295–303.
- Tarín, J.J.; Pérez-Albalá, S.; Cano, A. Consequences on offspring of abnormal function in ageing gametes. Hum. Reprod. Update 2000, 6, 532–549.
- Minson, C.T.; Wladkowski, S.L.; Cardell, A.F.; Pawelczyk, J.A.; Kenney, W.L. Age alters the cardiovascular response to direct passive heating. J. Appl. Physiol. 1998, 84, 1323–1332.
- Sagawa, S.; Shiraki, K.; Yousef, M.K.; Miki, K. Sweating and cardiovascular responses of aged men to heat exposure. J. Gerontol. 1988, 43, M1–M8.
- Kenney, W.; Morgan, A.; Farquhar, W.; Brooks, E.; Pierzga, J.M.; Derr, J. Decreased active vasodilator sensitivity in aged skin. Am. J. Physiol. Heart Circ. Physiol. 1997, 272, H1609–H1614.
- Kenney, W.L. Control of heat-induced cutaneous vasodilatation in relation to age. Eur. J. Appl. Physiol. Occup. Physiol. 1988, 57, 120–125.
- Martin, H.L.; Loomis, J.L.; Kenney, W.L. Maximal skin vascular conductance in subjects aged 5–85 yr. Appl. Physiol. 1995, 79, 297–301.
- Roddie, I.C.; Shepherd, J.T.; Whelan, R.F. The contribution of constrictor and dilator nerves to the skin vasodilatation during body heating. J. Physiol. 1957, 136, 489–497.
- Shastry, S.; Dietz, N.M.; Halliwill, J.R.; Reed, A.S.; Joyner, M.J. Effects of nitric oxide synthase inhibition on cutaneous vasodilation during body heating in humans. J. Appl. Physiol. 1998, 85, 830–834.
- Minson, C.T.; Holowatz, L.A.; Wong, B.J.; Kenney, W.L.; Wilkins, B.W. Decreased nitric oxide- and axon reflex-mediated cutaneous vasodilation with age during local heating. J. Appl. Physiol. 2002, 93, 1644–1649.
- Holowatz, L.A.; Houghton, B.L.; Wong, B.J.; Wilkins, B.W.; Harding, A.W.; Kenney, W.L.; Minson, C.T. Nitric oxide and attenuated reflex cutaneous vasodilation in aged skin. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1662–H1667.
- Bruning, R.S.; Santhanam, L.; Stanhewicz, A.E.; Smith, C.J.; Berkowitz, D.E.; Kenney, W.L.; Holowatz, L.A. Endothelial nitric oxide synthase mediates cutaneous vasodilation during local heating and is attenuated in middle-aged human skin. J. Appl. Physiol. 2012, 112, 2019–2026.
- Settelmeier, S.; Rassaf, T.; Hendgen-Cotta, U.B.; Stoffels, I. Nitric oxide generating formulation as an innovative approach to topical skin care: An open-label pilot study. Cosmetics 2021, 8, 16.
- Reckelhoff, J.F.; Kellum, J.A.; Blanchard, E.J.; Bacon, E.E.; Wesley, A.J.; Kruckeberg, W.C. Changes in nitric oxide precursor, l-arginine, and metabolites, nitrate and nitrite, with aging. Life Sci. 1994, 55, 1895–1902.
- Erdely, A.; Greenfeld, Z.; Wagner, L.; Baylis, C. Sexual dimorphism in the aging kidney: Effects on injury and nitric oxide system. Kidney Int. 2003, 63, 1021–1026.
- Baylis, C. Changes in renal hemodynamics and structure in the aging kidney; sexual dimorphism and the nitric oxide system. Exp. Gerontol. 2005, 40, 271–278.
- Doleželová, Š.; Jíchová, Š.; Husková, Z.; Vojtíšková, A.; Kujal, P.; Hošková, L.; Kautzner, J.; Sadowski, J.; Červenka, L.; Kopkan, L. Progression of hypertension and kidney disease in aging fawn-hooded rats is mediated by enhanced influence of renin-angiotensin system and suppression of nitric oxide system and epoxyeicosanoids. Clin. Exp. Hypertens. 2016, 38, 644–651.
- Vavrinec, P.; Henning, R.H.; Goris, M.; Landheer, S.W.; Buikema, H.; van Dokkum, R.P. Renal myogenic constriction protects the kidney from age-related hypertensive renal damage in the Fawn-Hooded rat. J. Hypertens. 2013, 31, 1637–1645.
- Ochodnický, P.; Henning, R.H.; Buikema, H.J.; de Zeeuw, D.; Provoost, A.P.; van Dokkum, R.P. Renal vascular dysfunction precedes the development of renal damage in the hypertensive Fawn-Hooded rat. Am. J. Physiol. Ren. Physiol. 2010, 298, F625–F633.
- Satoh, M.; Kidokoro, K.; Ozeki, M.; Nagasu, H.; Nishi, Y.; Ihoriya, C.; Fujimoto, S.; Sasaki, T.; Kashihara, N. Angiostatin production increases in response to decreased nitric oxide in aging rat kidney. Lab. Investig. J. Tech. Methods Pathol. 2013, 93, 334–343.
- Sarati, L.I.; Toblli, J.E.; Martinez, C.R.; Uceda, A.; Feldman, M.; Balaszczuk, A.M.; Fellet, A.L. Nitric oxide and AQP2 in hypothyroid rats: A link between aging and water homeostasis. Metab. Clin. Exp. 2013, 62, 1287–1295.
- Ferrer, J.E.; Velez, J.D.; Herrera, A.M. Age-related morphological changes in smooth muscle and collagen content in human corpus cavernosum. J. Sex. Med. 2010, 7, 2723–2728.
- Keegan, K.A.; Penson, D.F. Chapter 28—Vasculogenic Erectile Dysfunction. In Vascular Medicine: A Companion to Braunwald’s Heart Disease, 2nd ed.; Creager, M.A., Beckman, J.A., Loscalzo, J., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2013; pp. 341–348.
- El-Sakka, A.I. Reversion of penile fibrosis: Current information and a new horizon. Arab. J. Urol. 2011, 9, 49–55.
- Ferrini, M.G.; Davila, H.H.; Valente, E.G.; Gonzalez-Cadavid, N.F.; Rajfer, J. Aging-related induction of inducible nitric oxide synthase is vasculo-protective to the arterial media. Cardiovasc. Res. 2004, 61, 796–805.
This entry is offline, you can click here to edit this entry!