Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Chemistry, Applied
The abundant and inexpensive carbon monoxide (CO) is widely exploited as a C1 source for the synthesis of both fine and bulk chemicals. In this context, photochemical carbonylation reactions have emerged as a powerful tool for the sustainable synthesis of carbonyl-containing compounds (esters, amides, ketones, etc.).
- carbon monoxide
- carbonylation
- photochemical reactions
- visible light
- carbonyl-containing compounds
1. Introduction
Carbon monoxide (CO) is largely used in the chemical industry for manufacturing bulk chemicals (i.e., methanol, acetic acid) and fine chemicals (i.e., ibuprofen) and is frequently employed as an inexpensive and readily available C1 source in a wide range of carbonylative transformations for the synthesis of high-value-added carbonyl-containing compounds, such as acids, esters, amides and ketones [1][2][3][4][5][6]. The carbonyl unit is ubiquitous in a myriad of bioactive molecules, such as natural products, pharmaceuticals and agrochemicals, as well as materials. It can be also manipulated and transformed into a series of other functional groups, including amines, alcohols and olefins. In the era of sustainability, the development of economically improved and environmentally friendly catalytic protocols is highly recommended. In recent years, visible-light photocatalysis has received much attention from the synthetic chemists’ community since, unlike traditional thermal and catalytic reactions, it enables unprecedented reaction pathways, high selectivities and mild reaction conditions [7][8][9][10]. The combination of carbon monoxide-based carbonylation strategy with the advantages that come from the application of photocatalysis can potentially lead to a highly sustainable process [11][12][13][14][15]. However, the first reports displayed problems connected with a limited generality, poor selectivity and/or efficiency, high pressure of carbon monoxide or high energy of light irradiation [16][17][18][19][20][21][22][23]. Over the last two decades, new and more performing catalytic systems allowed to solve in part these issues. The major advances in the area of visible light-promoted carbonylations are here highlighted.
2 Visible-Light-Promoted Iridium-Catalyzed Carbonylations
Amides are ubiquitous in both natural and synthetic compounds, including pharmaceutical products. Odell and co-workers have recently developed a visible light-mediated fac-Ir(ppy)3 catalyzed amino carbonylation of unactivated alkyl iodides under mild reaction conditions [24]. The desired amides are produced in moderate to excellent yields through a two-chamber system (H-tube), where the CO is released ex situ from Mo(CO)6) (Scheme 1). Secondary and tertiary iodides can be successfully aminocarbonylated with a wide range of amine nucleophiles, whereas primary iodides provide satisfactory results in combination with hindered amines only. Remarkably, a dealkylative-aminocarbonylation pathway occurs when alkyl halides react with tertiary amines. The reaction mechanism starts with the reductive dehalogenation of an alkyl halide (R–X) to give the alkyl radical I, followed by CO insertion with the formation of an acyl radical intermediate II. The latter is either quenched by the starting alkyl iodide to produce an acyl iodide III or oxidized to an acylium ion IV. The nucleophilic attack of the amine on the acyl iodide or the acylium ion affords the final amide (Scheme 2).
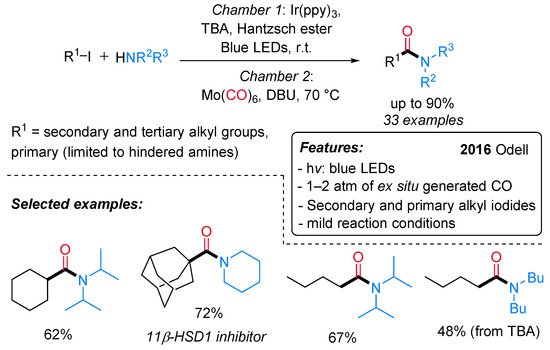
Scheme 1. Visible light-mediated fac-Ir(ppy)3-catalyzed aminocarbonylation of alkyl iodides.
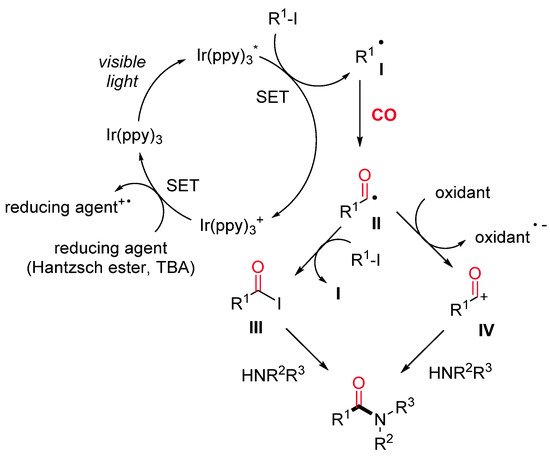
Scheme 2. The proposed reaction pathway.
A variety of acetate-containing 2,3-dihydrobenzofurans have been synthesized by Polyzos et al. through a visible light-mediated Ir-catalyzed radical carbonylation process under continuous flow conditions [25]. Overcoming the traditional limitations of both Pd-catalyzed alkoxycarbonylation reactions and classical radical-free carbonylation processes in terms of regioselectivity, the pressure of CO and sustainability [26], alkenyl-tethered arenediazonium salts underwent a versatile and stereoselective (exclusive 5-exo-dig cyclization) cyclization and alkoxycarbonylation cascade to a wide range of 2,3-dihydrobenzofuran derivatives in the presence of [Ir(dtbbpy)(ppy)2]PF6 as catalyst under blue LEDs (14 W) at room temperature (Scheme 3). Electron-donating and electron-withdrawing substituents on the arenediazonium salt moiety were nicely tolerated under the standard conditions. Regardless of steric bulk, alkyl alcohols were successfully employed as coupling partners to produce the desired esters in moderate to good yields. This continuous flow methodology also features a very short reaction time (200 s), moderate CO pressures (25 atm) and a straightforward scale-up. Interestingly, the commercial Ru(bpy)3Cl2·6H2O was found to be slightly less efficient in this transformation.
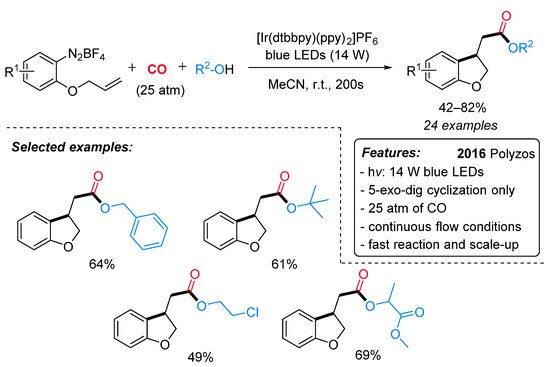
Scheme 3. Visible light-mediated [Ir(dtbbpy)(ppy)2]PF6-catalyzed cyclizative alkoxycarbonylation of alkenyl-tethered arenediazonium salts under continuous flow conditions.
The mechanism is supposed to start with the homolytic cleavage of the C–N bond of the allyloxy-tethered arenediazonium salt by single-electron transfer from the photocatalyst in its excited state (PC*), with the generation of an aryl radical I and the oxidized photocatalyst PC•+ (Scheme 4). An intramolecular radical alkene addition provides the primary alkyl radical II, which, after CO insertion, gives the acyl radical III. Oxidation of the acyl radical by the oxidized photocatalyst PC•+ results in the formation of an acylium ion IV and the regeneration of the photocatalyst PC. Lastly, the reaction of IV with alcohol leads to the final product.
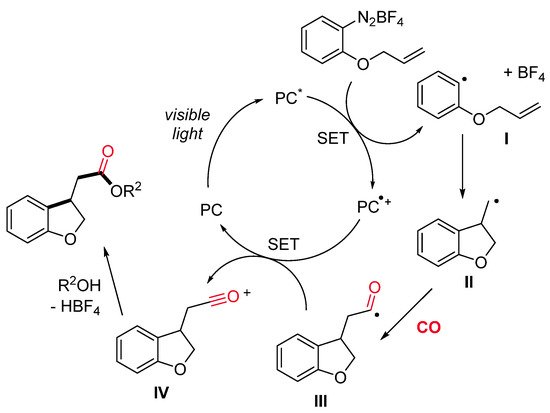
Scheme 4. A plausible reaction pathway.
In 2020, Polyzos and co-workers reported an Ir-catalyzed visible-light-promoted aminocarbonylation of alkyl and aryl halides with CO and amines under continuous flow conditions (Scheme 5) [27]. The iridium-based photocatalyst ([Ir(ppy)2(dtbbpy)]PF6) was used in combination with DIPEA (N,N-diisopropylethylamine) as a sacrificial reductant and under blue LED (54 W) irradiation. A large variety of aryl halides (I, Br, Cl) and alkyl iodides (primary, secondary and tertiary) reacted successfully with CO and an array of alkyl and aryl amines to provide the expected amides in good to high yields. The continuous flow system was assembled from commercially available components, featuring operational simplicity, high applicability, improved safety and easy scalability.
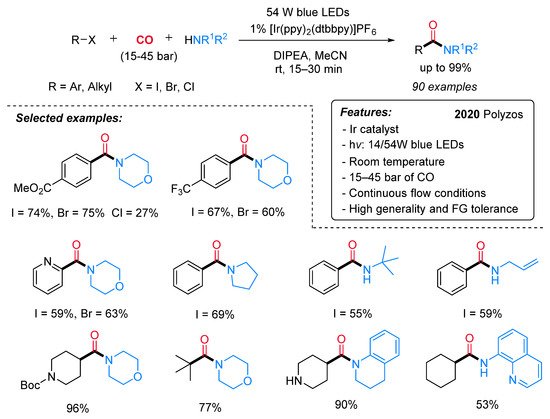
Scheme 5. Visible light-mediated [Ir(dtbbpy)(ppy)2]PF6-catalyzed aminocarbonylation of aryl and alkyl halides under continuous flow conditions.
Ynones represent an important motif present in bioactive compounds and are useful intermediates in the synthesis of heterocycles [28]. Alternative to the previously described Ryu’s method [29], Lu and Xiao et al. have recently developed an efficient protocol for the synthesis of ynones through a visible-light-induced photocatalytic decarboxylative carbonylative alkynylation of carboxylic acids in the presence of carbon monoxide (CO) at room temperature [30]. The decarboxylative alkynylation reaction was carried out using ethynylbenziodoxolones (EBX) as the alkynylating agent, carboxylic acid, in the presence of Ir[dF(CF3)ppy]2(dtbbpy)PF6 as a photocatalyst and Cs2CO3 as a base (Scheme 6). A range of aliphatic carboxylic acids gave the corresponding ynones in good to excellent yields under mild conditions but extremely high pressure of CO. Mechanistically, it is proposed that the excited photocatalyst Ir(III)*, generated from Ir(III) under visible-light irradiation, is able to oxidize the substrate with the formation of an alkyl radical I (Scheme 7). Then, the latter, after CO insertion, gives rise to an acyl radical II, which undergoes radical addition to the alkylating agent affording the radical intermediate III. The subsequent radical elimination reaction yields the desired ynone and the benziodoxolonyl radical IV. Finally, radical IV is reduced to ortho-iodobenzoate by Ir(II), which, in turn, is oxidized to Ir(III) for a new catalytic cycle.
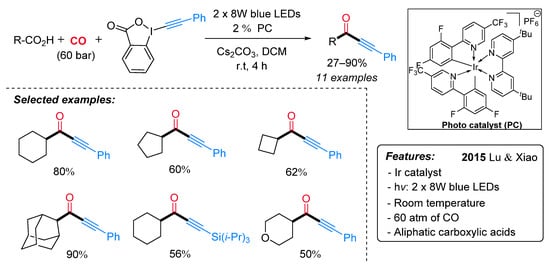
Scheme 6. Visible light-promoted photo-catalyzed decarboxylative carbonylative alkynylation of carboxylic acids to ynones.
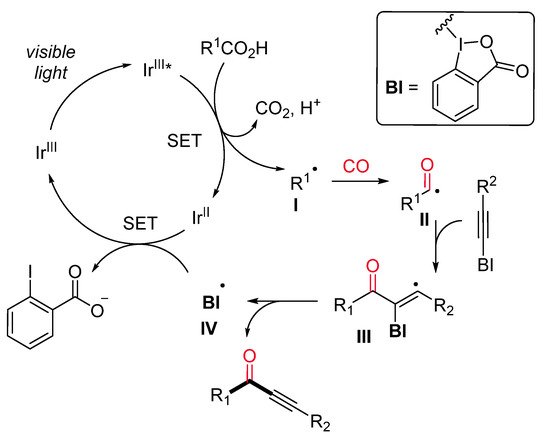
Scheme 7. The proposed reaction mechanism.
More recently, the same research group developed a visible-light-driven photocatalyzed synthesis of α,β-unsaturated ketones starting from alkyl Katritzky salts as a source of radicals [31]. In the context of a more general reactivity, a limited number of alkyl Katritzky salts reacted with 1,1-diphenylethylene in the presence of [Ir(4-Fppy)2(bpy)]PF6 and DABCO in MeCN under CO atmosphere and blue LEDs irradiation, leading to α,β-unsaturated ketones (Scheme 8). In the proposed reaction mechanism, the excited photocatalyst Ir(III)* is supposed to reduced alkyl Katritzky salts through a SET process (Scheme 9). The generated radicals I inserts one molecule of CO with the formation of an acyl radical II, which reacts with 1,1-diphenylethylene yielding the radical species III. Intermediate III is then reduced by the photocatalyst Ir(IV) with the formation of the cationic intermediate IV, which, lastly, undergoes a DABCO-mediated deprotonation step to deliver the final ketone.
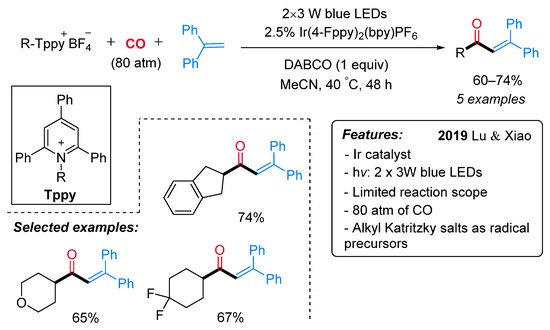
Scheme 8. Visible light-promoted Ir-catalyzed carbonylative alkyl-Heck-type reactions to α,β-unsaturated ketones.
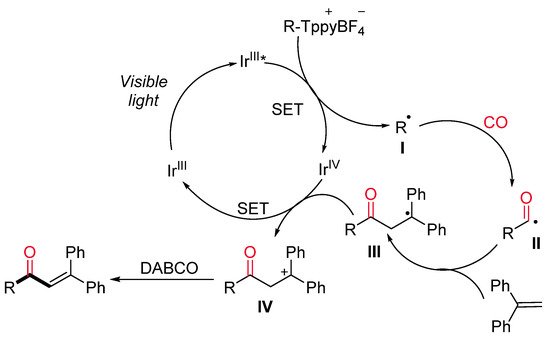
Scheme 9. The proposed reaction mechanism.
3. Visible-Light-Promoted Cobalt-Catalyzed Carbonylations
Cobalt salts have been found to efficiently catalyze the carbonylation of alkenes, chloro and bromoalkanes under UV irradiation [32][33][34][35][36]. The first report on visible-light-mediated cobalt-catalyzed carbonylation reaction appeared in 2012, when Jia, Yin and colleagues, based on their previous findings [37], developed a Co(OAc)2-catalyzed alkoxycarbonylation of aryl bromides under visible-light irradiation and mild conditions (Scheme 10) [38]. The use of a strong base (NaOMe) and an organic sensitizer (PhCOPh) was essential to the reaction’s success. Aryl bromides with a chloro substituent behaved better than tolyl bromides; however, a selectivity higher than 99% was always observed.
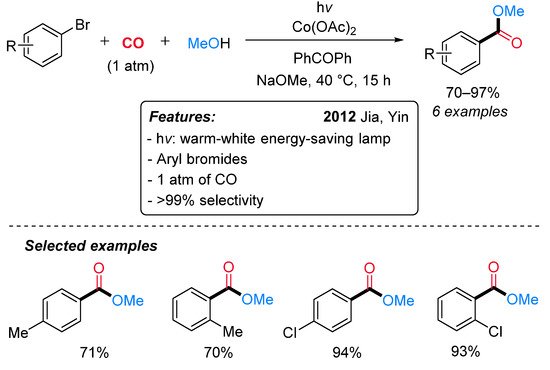
Scheme 10. Visible-light-mediated cobalt-catalyzed alkoxycarbonylation of aryl bromides.
Recently, Alexanian et al. reported the visible-light-promoted aminocarbonylation of (hetero) aryl/vinyl bromides and chlorides using an inexpensive cobalt catalyst (Co2(CO)8) in conjunction with tetramethylpiperidine (TMP) (Scheme 11) [39].
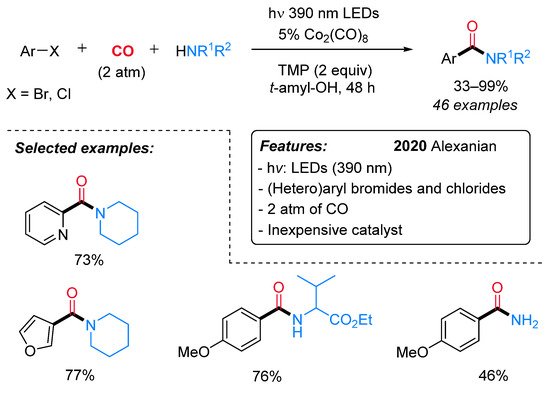
Scheme 11. Visible-light-mediated cobalt-catalyzed aminocarbonylation of bromo and chloroaryls.
Aryl bromides having both electron-rich and electron-deficient groups, as well as vinyl bromides, were well tolerated. In addition, bromo heteroaryls, such as pyridines, furans, quinolines and indoles, also performed nicely in this reaction. Aryl chlorides with electron-deficient groups gave the expected amides in satisfactory to good yields. A wide variety of primary and secondary aliphatic amines, including amino acids, gave the corresponding amides in moderate to high yields. Preliminary mechanistic investigations were consistent with the first formation of a cobaltate anion [Co(CO)4]− (I) as an active catalyst and the subsequent generation of an electron donor–acceptor complex (EDA) II (Scheme 12). The intermediacy of the visible light leads to the corresponding excited species III. Then, a single-electron transfer produces two radical species (IV), which recombines, leading to a (hetero)aryl or vinyl cobalt species V. Finally, migratory insertion of CO affords intermediate VI, which undergoes displacement of the amine delivering the amide and the active catalyst.
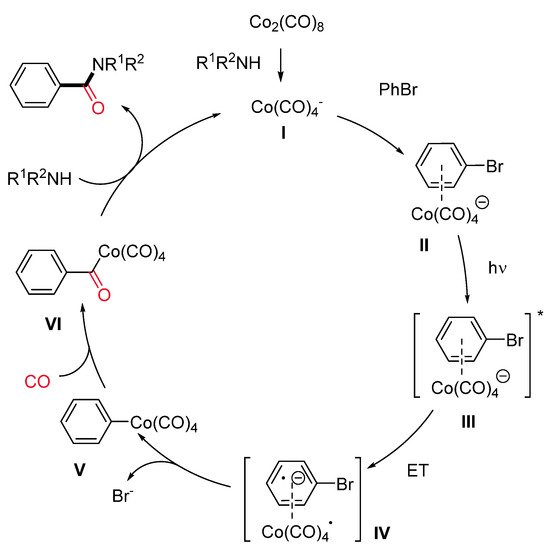
Scheme 12. The proposed reaction pathway.
4. Visible-Light-Promoted Rutenium-Catalyzed Carbonylations
Carboxylic acids are ubiquitous in a wide range of bioactive molecules and industrially relevant compounds and represent a versatile, functional group in organic synthesis. Bousquet et al. have recently reported a ruthenium-catalyzed visible-light-promoted synthesis of benzoic acids starting from aryl diazonium salts, carbon monoxide (10–50 atm) and water under mild conditions (Scheme 13) [40]. The reaction displays a good functional group tolerance and generality. Notably, satisfactory results were achieved directly starting from anilines through the in situ generation of aryl diazonium salts, avoiding in this way their isolation. The proposed reaction pathway starts with the generation of an aryl radical (I) by SET with the excited Ru(II)* (Scheme 14). Next, the insertion of CO on the aryl radical gives rise to an acyl radical (II), which is oxidized by the Ru(III) species to an acylium ion III. The Ru(II) is regenerated, and the acylium ion reacts with water delivering the desired carboxylic acid.
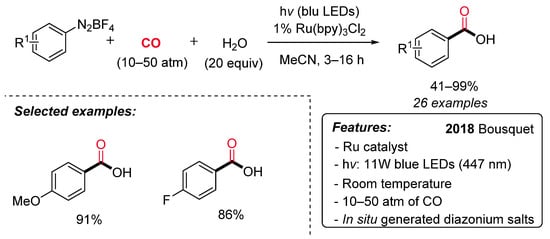
Scheme 13. The Ru-catalyzed synthesis of carboxylic acids via hydroxycarbonylation of aryl diazonium salts under irradiation conditions.
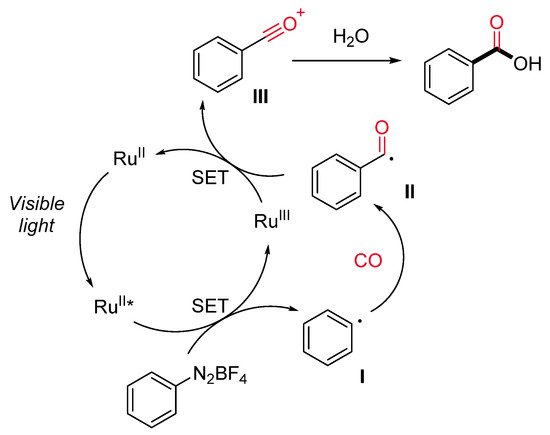
Scheme 14. The proposed reaction mechanism for the hydroxycarbonylation reaction.
5. Visible-Light-Promoted Copper-Catalyzed Carbonylations
To the best of our knowledge, the first and only example of copper-catalyzed photoinduced carbonylation reaction has been recently reported by Chen and co-workers [41] and is aimed at the synthesis of cyano-tethered amides by aminocarbonylation of oxime esters with amines and CO under mild conditions (Scheme 15). A number of cycloketone oxime esters and alkyl/aryl amines were evaluated, and a high level of functional group tolerance was found. Larger than four-membered rings, such as cyclopentanone and cyclohexanone oxime esters, did not lead to the expected products. For this aminocarbonylation reaction, the authors proposed a visible light-mediated Cu(I)/Cu(II)/Cu(III)-based catalytic cycle (Scheme 16). The first step is a single-electron transfer between the photoexcited LnCu(I)–NHPh complex (II)* or, alternatively, the ground state LnCu(I)–NHPh species (I) and the oxime, leading to an iminyl radical III and the oxidized LnCu(II)–NHPh complex (IV). Then, III undergoes a selective β–C–C bond cleavage to form the alkyl radical V, which can react with Cu(II)-complex IV producing a Cu(III) organometallic species VI. The latter, after sequential coordination and insertion of CO, leads to an acyl copper intermediate (VII or VIII), providing the final amide by reductive elimination and regenerating the active Cu(I) catalyst.
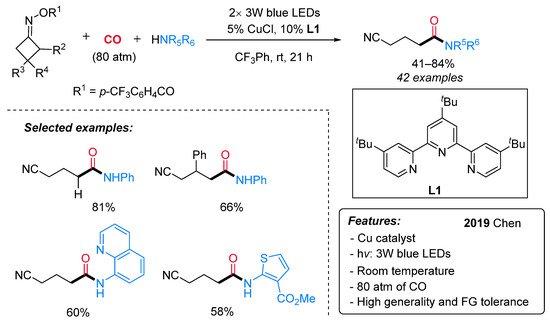
Scheme 15. The Cu-catalyzed synthesis of amides via aminocarbonylation of oximes under visible-light irradiation.
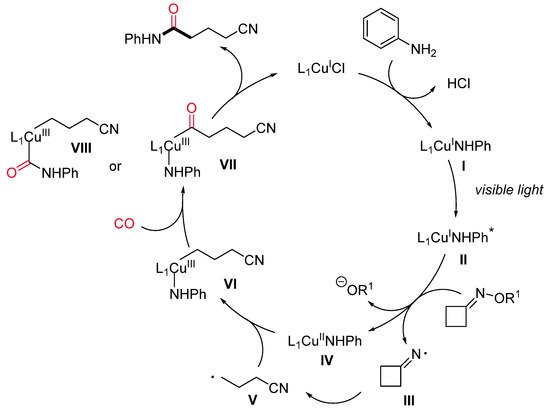
Scheme 16. The proposed reaction pathway.
6. Visible-Light-Promoted Metal-Free Carbonylations
The major achievements in visible-light-mediated carbonylation reactions were obtained under transition-metal catalysis. The requirement of transition-metal catalysts often in connection with organic ligands is undoubtedly expensive, and the removal of their traces from the final products is necessary, particularly in the synthesis of pharmaceutical compounds. Metal-free photocatalytic protocols can overcome the above-mentioned drawbacks and may represent attractive greener and more sustainable alternatives.
In 1997, the efficient conversion of alkyl iodides to the corresponding esters was achieved under photoirradiation conditions in the absence of catalysts (Scheme 17) [42]. The presence of hydrogens in the β position on the alkyl iodides results in the formation of positional isomers via the β-elimination pathway under conventional transition metal-catalyzed carbonylation conditions [43]. This radical protocol allows the selective alkoxycarbonylation at the carbon attached to the halogen, and, for this reason, a wide range of aliphatic iodides, including tertiary ones, can be efficiently employed (Scheme 17, selected examples). High pressure of CO (from 20 to 55 atm) and the presence of an organic (triethylamine) or inorganic base (K2CO3, KOH) were essential to the reaction outcome. Mechanistically, by irradiation, the homolytic cleavage of the C–I bond produces an alkyl radical species that reacts with the carbon monoxide, providing an acyl radical intermediate. In the presence of another molecule of alkyl iodide, an acyl iodide is then generated, and, after reaction with R2OH, the corresponding ester is produced (Scheme 17, proposed mechanism). Remarkably, the reaction can be extended to the synthesis of amides using amines in place of alcohols [44].
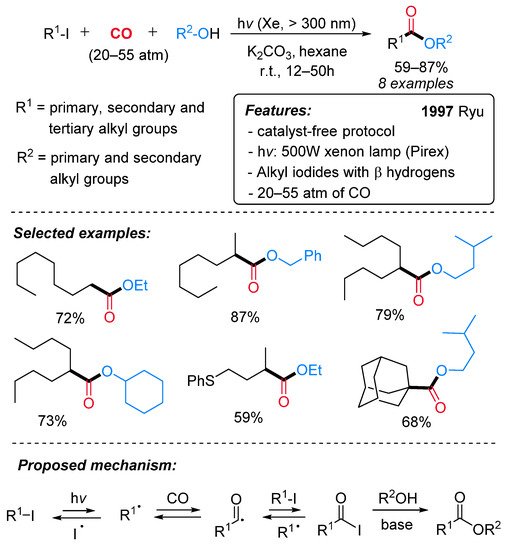
Scheme 17. The catalyst-free synthesis of esters via radical alkoxycarbonylation of alkyl iodides under irradiation conditions.
The first example of radical alkoxycarboxylation of aryldiazonium salts using CO gas through visible-light-induced photoredox catalysis has been reported in 2015 by the Xiao research group (Scheme 18) [45]. A wide variety of arylcarboxylic acid esters, bearing both electron-donating and electron-withdrawing groups, were obtained in good to excellent yields at room temperature. The generality of the alcohol coupling partner, including the use of chiral alcohols, was also demonstrated. Notably, terminal alkynes, which are known to be good acceptors in radical reactions, remained untouched. Sensitive functional groups (such as iodo, bromo) in traditional transition metal-catalyzed alkoxycarbonylation reactions were compatible in this process, thus allowing further synthetic manipulations. Remarkably, a low loading of an organic dye (fluorescein) is employed as a photocatalyst, and a low energy visible light (16 W blue LEDs) is enough to promote the reaction. However, high pressure of CO (80 atm) proved to be crucial for the success of the reaction.
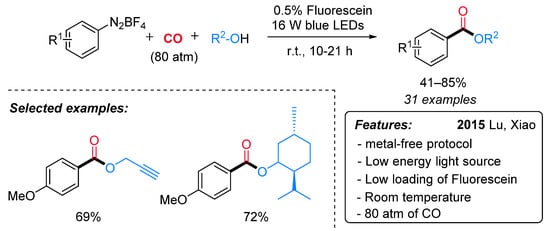
Scheme 18. Visible light-induced fluorescein-catalyzed alkoxycarboxylation of aryldiazonium salts.
This radical alkoxycarboxylation protocol was applicable to other radical carboxylation cascade reactions. For example, when benzenediazonium salts, bearing the allyl or the propargyl function at the ortho position, were caused to react under standard reaction conditions, the corresponding methyl 2-(2,3-dihydrobenzofuran-3-yl)acetate (Scheme 19a) and methyl 2-(benzofuran-3-yl)acetate (Scheme 19b) were obtained in satisfactory yields. Mechanistic studies suggested that the reaction might proceed via carbon radical intermediates I formed by single-electron reduction of the substrate with the fluorescein photocatalyst in its excited state. The reactive intermediate I might be able to trap the CO molecule delivering the benzoyl radical II, which is oxidized by the dye radical cation (Dye·+) with concomitant restoring of the active photocatalyst. The resulting cation III could be directly trapped by various alcohols leading to a wide range of alkyl benzoates (Scheme 20).
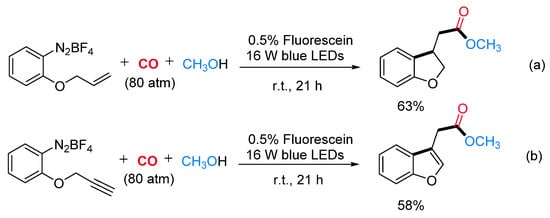
Scheme 19. Fluorescein-catalyzed radical addition/alkoxycarboxylation reaction sequences from alkene (a) and alkyne (b) derivatives.
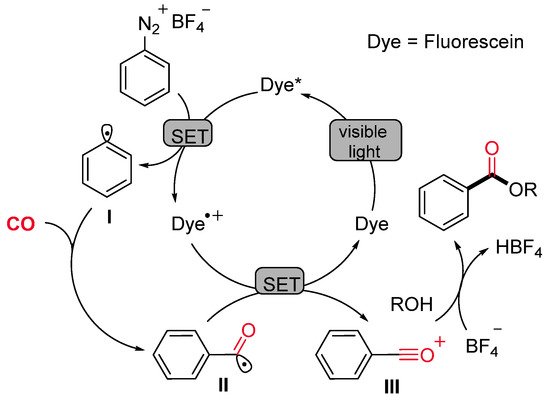
Scheme 20. The proposed mechanism for the fluorescein-catalyzed visible light-induced alkoxycarboxylation of aryldiazonium salts.
A very similar approach was developed in the same period by Jacobi von Wangelin’s group. Alkyl benzoates were efficiently obtained from arene diazonium salts, carbon monoxide and alcohols under visible-light irradiation and in the presence of eosin Y as the catalyst (Scheme 21) [46]. Under metal and base-free conditions, a variety of functionalities were well tolerated both on the substrates and on various functionalized additives. Interestingly, tertiary esters, which are difficult to obtain by conventional esterification procedures due to their steric hindrance, can also be prepared in excellent yields. Moreover, the industrially relevant intermediate, 2-ethylhexyl benzoate, has been obtained in 58% yield from PhN2BF4, 2-ethylhexanol and CO under standard conditions.
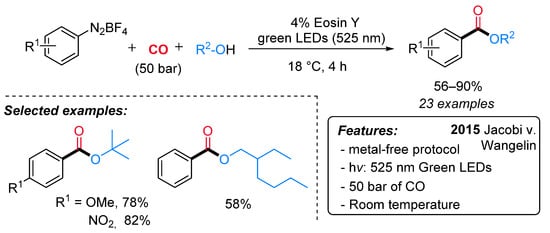
Scheme 21. The synthesis of benzoates through visible light-driven eosin Y-catalyzed alkoxycarbonylation of arene diazonium salts.
The mechanistic investigations, including the observation of benzoyl-TEMPO adduct, support the sequential reduction (SET), carbonylation, and oxidation (SET) to aroylium cations, which undergo rapid addition to alcohols (Scheme 20, Dye = Eosin Y).
From the pioneering work of Heck on palladium-catalyzed aminocarbonylation of aryl iodides [47], several efforts have been accomplished for the synthesis of aromatic amides [48][49] through radical carbonylative pathways as well [45][46][50]. Recently, the first example of catalyst-free photo-induced aminocarbonylation of aryl iodides with CO and amines has been reported by Ryu and co-workers [51]. A wide variety of amides, including hetero aromatic amides, has been obtained in good yields under mild reaction conditions (Scheme 22).
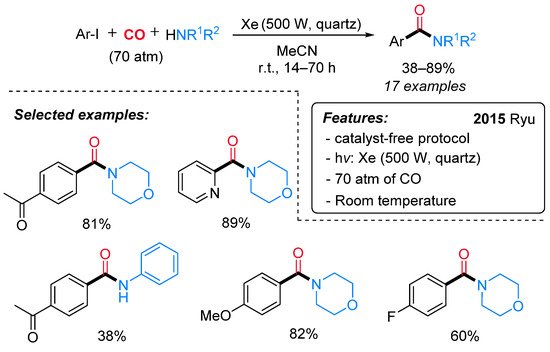
Scheme 22. Photo-induced aminocarbonylation of aryl iodides with primary and secondary amines.
The aryl radical I, generated by a photo-induced cleavage of the starting aryl iodide, might react with CO leading to the corresponding acyl radical II (Scheme 23). The nucleophilic addition of an amine to the latter gives a zwitterionic radical intermediate III. Finally, electron transfer to the aryl iodide would provide the aryl radical I and the expected amide.
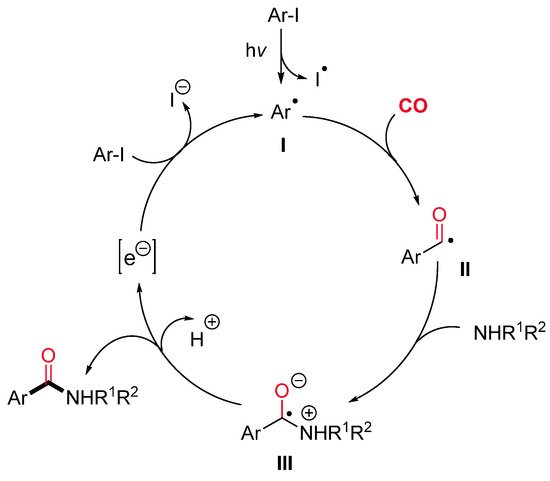
Scheme 23. The proposed mechanistic pathway for the catalyst-free photo-induced aminocarbonylation of aryl iodides.
Inspired by Xiao [45] and Wangelin [46] independent reports, the Eosin-Y-catalyzed synthesis of indol-3-yl aryl ketones from indoles, CO and aryldiazonium salts has been recently reported by Gu and Li (Scheme 24) [52]. This protocol features high versatility, high functional group tolerance and mild reaction conditions except for the high pressure of CO required (70 atm).
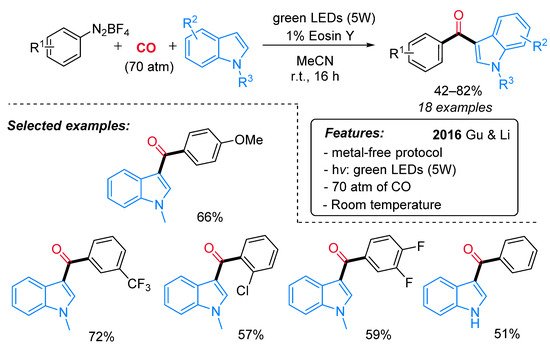
Scheme 24. The photo-induced synthesis of indol-3-yl aryl ketones from indoles, CO and aryldiazonium salts.
The synthesis of indol-3-yl aryl ketones was achieved also by Li, Liang and colleagues, starting from arylsulfonyl chlorides in place of aryldiazonium salts under very similar photocatalytic conditions (Scheme 25) [53]. Notably, the methodology demonstrated wide applicability with both electron-rich and electron-deficient functional groups at reduced reaction time if compared with Gu and Li’s protocol [52]. The proposed pathway starts with the reduction of the arylsulfonyl chloride by a single-electron transfer process (SET) to produce an aryl radical I and the Eosin radical cation [Eosin˙+] (Scheme 26). Then, the aryl radical I reacts with CO, delivering an acyl radical II, which is oxidized to an acylium intermediate III by the Eosin radical cation [Eosin·+]. The active photocatalytic species is indeed regenerated while the acylium ion undergoes nucleophilic attack by the indolyl species, leading to the final product.
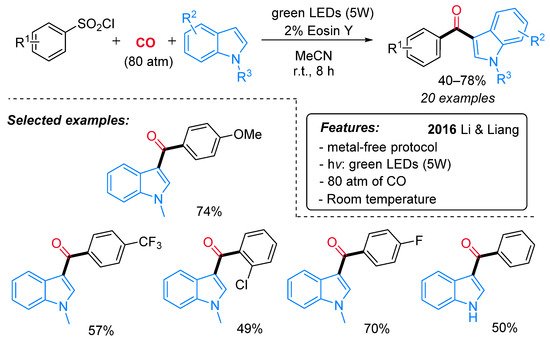
Scheme 25. The photo-induced synthesis of indol-3-yl aryl ketones from indoles, CO and aryl sulfonyl chlorides.
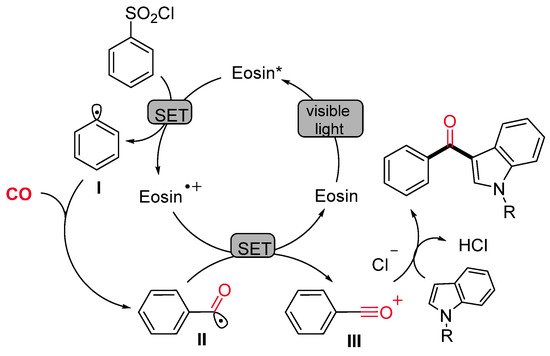
Scheme 26. The proposed mechanism for the Eosin-catalyzed visible-light-promoted indolylcarboxylation of aryl sulfonyl chlorides.
This entry is adapted from the peer-reviewed paper 10.3390/catal11080918
References
- Zhu, C.; Liu, J.; Li, M.-B.; Bäckvall, J.-E. Palladium-Catalyzed Oxidative Dehydrogenative Carbonylation Reactions Using Carbon Monoxide and Mechanistic Overviews. Chem. Soc. Rev. 2020, 49, 341–353.
- Yin, Z.; Xu, J.-X.; Wu, X.-F. No Making without Breaking: Nitrogen-Centered Carbonylation Reactions. ACS Catal. 2020, 10, 6510–6531.
- Zhang, S.; Neumann, H.; Beller, M. Synthesis of α,β-Unsaturated Carbonyl Compounds by Carbonylation Reactions. Chem. Soc. Rev. 2020, 49, 3187–3210.
- Peng, J.-B.; Geng, H.-Q.; Wu, X.-F. The Chemistry of CO: Carbonylation. Chemistry 2019, 5, 526–552.
- Peng, J.-B.; Wu, F.-P.; Wu, X.-F. First-Row Transition-Metal-Catalyzed Carbonylative Transformations of Carbon Electrophiles. Chem. Rev. 2019, 119, 2090–2127.
- Mancuso, R.; Della Ca’, N.; Veltri, L.; Ziccarelli, I.; Gabriele, B. PdI2-Based Catalysis for Carbonylation Reactions: A Personal Account. Catalysts 2019, 9, 610.
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166.
- Prier, C.K.; Rankic, D.A.; Mac-Millan, D.W. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363.
- Xuan, J.; Xiao, W.-J. Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2012, 51, 6828–6838.
- Narayanam, J.M.R.; Stephenson, C.R.J. Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev. 2011, 40, 102–113.
- Singh, J.; Sharma, S.; Sharma, A. Photocatalytic Carbonylation Strategies: A Recent Trend in Organic Synthesis. J. Org. Chem. 2021, 86, 24–48.
- Hu, X.-Q.; Liu, Z.-K.; Xiao, W.-J. Radical Carbonylative Synthesis of Heterocycles by Visible Light Photoredox Catalysis. Catalysts 2020, 10, 1054.
- Zhao, S.; Mankad, N.P. Metal-Catalysed Radical Carbonylation Reactions. Catal. Sci. Technol. 2019, 9, 3603–3613.
- Peng, J.-B.; Qi, X.; Wu, X.-F. Visible Light-Induced Carbonylation Reactions with Organic Dyes as the Photosensitizers. ChemSusChem 2016, 9, 2279–2283.
- Sumino, S.; Fusano, A.; Fukuyama, T.; Ryu, I. Carbonylation Reactions of Alkyl Iodides through the Interplay of Carbon Radicals and Pd Catalysts. Acc. Chem. Res. 2014, 47, 1563–1574.
- Kunin, A.J.; Eisenberg, R. Photochemical Carbonylation of Benzene by Iridium(I) and Rhodium(I) Square-Planar Complexes. Organometallics 1988, 7, 2124–2129.
- Ferguson, R.R.; Crabtree, R.H. Mercury-Photosensitized Sulfination, Hydrosulfination, and Carbonylation of Hydrocarbons: Alkane and Alkene Conversion to Sulfonic Acids, Ketones, and Aldehydes. J. Org. Chem. 1991, 56, 5503–5510.
- Sakakura, T.; Sodeyama, T.; Sasaki, K.; Wada, K.; Tanaka, M. Carbonylation of Hydrocarbons via Carbon-Hydrogen Activation Catalyzed by RhCl(CO)(PMe3)2 under Irradiation. J. Am. Chem. Soc. 1990, 112, 7221–7229.
- Jaynes, B.S.; Hill, C.L. Radical Carbonylation of Alkanes via Polyoxotungstate Photocatalysis. J. Am. Chem. Soc. 1995, 117, 4704–4705.
- Ryu, I.; Muraoka, H.; Kambe, N.; Komatsu, M.; Sonoda, N. Group Transfer Carbonylations: Photoinduced Alkylative Carbonylation of Alkenes Accompanied by Phenylselenenyl Transfer. J. Org. Chem. 1996, 61, 6396–6403.
- Pagenkopf, B.L.; Livinghouse, T. Photochemical Promotion of the Intramolecular Pauson−Khand Reaction. A New Experimental Protocol for Cobalt-Catalyzed [2 + 2 + 1] Cycloadditions. J. Am. Chem. Soc. 1996, 118, 2285–2286.
- Ogawa, A.; Sumino, Y.; Nanke, T.; Ohya, S.; Sonoda, N.; Hirao, T. Photoinduced Reduction and Carbonylation of Organic Chlorides with Samarium Diiodide. J. Am. Chem. Soc. 1997, 119, 2745–2746.
- Sigman, M.S.; Eaton, B.E. The First Iron-Mediated Catalytic Carbon-Nitrogen Bond Formation: [4 + 1] Cycloaddition of Allenyl Imines and Carbon Monoxide. J. Org. Chem. 1994, 59, 7488–7491.
- Chow, S.Y.; Stevens, M.Y.; Åkerbladh, L.; Bergman, S.; Odell, L.R. Mild and Low-Pressure fac-Ir(Ppy)3 -Mediated Radical Aminocarbonylation of Unactivated Alkyl Iodides through Visible-Light Photoredox Catalysis. Chem. Eur. J. 2016, 22, 9155–9161.
- Micic, N.; Polyzos, A. Radical Carbonylation Mediated by Continuous-Flow Visible-Light Photocatalysis: Access to 2,3-Dihydrobenzofurans. Org. Lett. 2018, 20, 4663–4666.
- Ryu, I.; Niguma, T.; Minakata, S.; Komatsu, M.; Hadida, S.; Curran, D.P. Hydroxymethylation of Organic Halides. Evaluation of a Catalytic System Involving a Fluorous Tin Hydride Reagent for Radical Carbonylation. Tetrahedron Lett. 1997, 38, 7883–7886.
- Forni, J.A.; Micic, N.; Connell, T.U.; Weragoda, G.; Polyzos, A. Tandem Photoredox Catalysis: Enabling Carbonylative Amidation of Aryl and Alkylhalides. Angew. Chem. Int. Ed. 2020, 59, 18646–18654.
- Wang, Z.; Li, L.; Huang, Y. A General Synthesis of Ynones from Aldehydes via Oxidative C–C Bond Cleavage under Aerobic Conditions. J. Am. Chem. Soc. 2014, 136, 12233–12236.
- Fusano, A.; Fukuyama, T.; Nishitani, S.; Inouye, T.; Ryu, I. Synthesis of Alkyl Alkynyl Ketones by Pd/Light-Induced Three-Component Coupling Reactions of Iodoalkanes, CO, and 1-Alkynes. Org. Lett. 2010, 12, 2410–2413.
- Zhou, Q.-Q.; Guo, W.; Ding, W.; Wu, X.; Chen, X.; Lu, L.-Q.; Xiao, W.-J. Decarboxylative Alkynylation and Carbonylative Alkynylation of Carboxylic Acids Enabled by Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2015, 54, 11196–11199.
- Jiang, X.; Zhang, M.; Xiong, W.; Lu, L.; Xiao, W. Deaminative (Carbonylative) Alkyl-Heck-type Reactions Enabled by Photocatalytic C−N Bond Activation. Angew. Chem. Int. Ed. 2019, 58, 2402–2406.
- Brunet, J.-J.; Sidot, C.; Caubere, P. Cobalt Carbonyl Catalyzed SRN1 Carbonylation of Aryl and Vinyl Halides by Phase Transfer Catalysis. Tetrahedron Lett. 1981, 22, 1013–1016.
- Brunet, J.J.; Sidot, C.; Caubere, P. Sunlamp-Irradiated Phase-Transfer Catalysis. 1. Cobalt Carbonyl Catalyzed SRN1 Carbonylations of Aryl and Vinyl Halides. J. Org. Chem. 1983, 48, 1166–1171.
- Dragojlovic, V.; Gao, D.B.; Chow, Y.L. Multigram Scale Cobalt Catalyzed Photochemical Methoxycarbonylation of Alkenes. J. Mol. Catal. Chem. 2001, 171, 43–51.
- Cash, D.; Combs, A.; Dragojlovic, V. Cobalt-Catalyzed Photolytic Methoxycarbonylation of Bromoalkanes in the Presence of a Lewis Acid. Tetrahedron Lett. 2004, 45, 1143–1145.
- Jia, Y.P.; Cui, Y.N.; Yin, J.M.; Zhou, G.Y.; Li, S.M.; Gao, D.B.; Wang, X.S. Cobalt-Catalyzed Photopromoted Carbonylation of Chloroalkanes in the Presence of KI. Chin. Chem. Lett. 2010, 21, 1033–1036.
- Mu, L.Z.; Jia, Y.P.; Yin, J.M.; Zhou, G.Y.; Cui, Y.N.; Gao, D.B. Photopromoted Carbonylation of Bromobenzene under Ambient Conditions. Chin. Chem. Lett. 2009, 20, 531–534.
- Zhong, W.H.; Cui, Y.N.; Li, S.M.; Jia, Y.P.; Yin, J.M. The Carbonylation of Phenyl Bromide and Its Derivatives under Visible Light Irradiation. Chin. Chem. Lett. 2012, 23, 29–32.
- Veatch, A.M.; Alexanian, E.J. Cobalt-Catalyzed Aminocarbonylation of (Hetero)Aryl Halides Promoted by Visible Light. Chem. Sci. 2020, 11, 7210–7213.
- Gosset, C.; Pellegrini, S.; Jooris, R.; Bousquet, T.; Pelinski, L. Visible-Light-Mediated Hydroxycarbonylation of Diazonium Salts. Adv. Synth. Catal. 2018, 360, 3401–3405.
- Lu, B.; Cheng, Y.; Chen, L.-Y.; Chen, J.-R.; Xiao, W.-J. Photoinduced Copper-Catalyzed Radical Aminocarbonylation of Cycloketone Oxime Esters. ACS Catal. 2019, 9, 8159–8164.
- Nagahara, K.; Ryu, I.; Komatsu, M.; Sonoda, N. Radical Carboxylation: Ester Synthesis from Alkyl Iodides, Carbon Monoxide, and Alcohols under Irradiation Conditions. J. Am. Chem. Soc. 1997, 119, 5465–5466.
- Urata, H.; Maekawa, H.; Takahashi, S.; Fuchikami, T. Transition Metal Complex-Catalyzed Carbonylation of Organic Halides in N,N,N’,N’-Tetraalkylurea Solution in the Absence of Added Base. J. Org. Chem. 1991, 56, 4320–4322.
- Ryu, I. Radical Carboxylations of Iodoalkanes and Saturated Alcohols Using Carbon Monoxide. Chem. Soc. Rev. 2001, 30, 16–25.
- Guo, W.; Lu, L.-Q.; Wang, Y.; Wang, Y.-N.; Chen, J.-R.; Xiao, W.-J. Metal-Free, Room-Temperature, Radical Alkoxycarbonylation of Aryldiazonium Salts through Visible-Light Photoredox Catalysis. Angew. Chem. Int. Ed. 2015, 54, 2265–2269.
- Majek, M.; Jacobi von Wangelin, A. Metal-Free Carbonylations by Photoredox Catalysis. Angew. Chem. Int. Ed. 2015, 54, 2270–2274.
- Schoenberg, A.; Heck, R.F. Palladium-Catalyzed Amidation of Aryl, Heterocyclic, and Vinylic Halides. J. Org. Chem. 1974, 39, 3327–3331.
- Friis, S.D.; Skrydstrup, T.; Buchwald, S.L. Mild Pd-Catalyzed Aminocarbonylation of (Hetero)Aryl Bromides with a Palladacycle Precatalyst. Org. Lett. 2014, 16, 4296–4299.
- Xu, T.; Alper, H. Pd-Catalyzed Chemoselective Carbonylation of Aminophenols with Iodoarenes: Alkoxycarbonylation vs Aminocarbonylation. J. Am. Chem. Soc. 2014, 136, 16970–16973.
- Kawamoto, T.; Okada, T.; Curran, D.P.; Ryu, I. Efficient Hydroxymethylation Reactions of Iodoarenes Using CO and 1,3-Dimethylimidazol-2-Ylidene Borane. Org. Lett. 2013, 15, 2144–2147.
- Kawamoto, T.; Sato, A.; Ryu, I. Photoinduced Aminocarbonylation of Aryl Iodides. Chem. Eur. J. 2015, 21, 14764–14767.
- Zhang, H.-T.; Gu, L.-J.; Huang, X.-Z.; Wang, R.; Jin, C.; Li, G.-P. Synthesis of Indol-3-Yl Aryl Ketones through Visible-Light-Mediated Carbonylation. Chin. Chem. Lett. 2016, 27, 256–260.
- Li, X.; Liang, D.; Huang, W.; Zhou, H.; Li, Z.; Wang, B.; Ma, Y.; Wang, H. Visible Light-Induced Carbonylation of Indoles with Arylsulfonyl Chlorides and CO. Tetrahedron 2016, 72, 8442–8448.
This entry is offline, you can click here to edit this entry!