Cancer cachexia is a syndrome experienced by many patients with cancer. Exercise can act as an autophagy modulator, and thus holds the potential to be used to treat cancer cachexia. Autophagy imbalance plays an important role in cancer cachexia, and is correlated to skeletal and cardiac muscle atrophy and energy-wasting in the liver. The molecular mechanism of autophagy modulation in different types of exercise has not yet been clearly defined. This review aims to elaborate on the role of exercise in modulating autophagy in cancer cachexia.
1. Overview
Cancer cachexia is a syndrome experienced by many patients with cancer. Exercise can act as an autophagy modulator, and thus holds the potential to be used to treat cancer cachexia. Autophagy imbalance plays an important role in cancer cachexia, and is correlated to skeletal and cardiac muscle atrophy and energy-wasting in the liver. The molecular mechanism of autophagy modulation in different types of exercise has not yet been clearly defined. This review aims to elaborate on the role of exercise in modulating autophagy in cancer cachexia. We evaluated nine studies in the literature and found a potential correlation between the type of exercise and autophagy modulation. Combined exercise or aerobic exercise alone seems more beneficial than resistance exercise alone in cancer cachexia. Looking ahead, determining the physiological role of autophagy modulated by exercise will support the development of a new medical approach for treating cancer cachexia. In addition, the harmonization of the exercise type, intensity, and duration might play a key role in optimizing the autophagy levels to preserve muscle function and regulate energy utilization in the liver.
2. Cachexia
Patients with cancer usually experience cachexia; cancer cachexia is a multifactorial syndrome correlated with cancer, characterized by skeletal muscle decline that cannot be fully recovered by nutritional support, and eventually leads to dysfunction of the muscle [
1,
2]. This syndrome manifests in clinical symptoms, such as weight loss, muscle atrophy, anorexia, fatigue, anemia, and edema, which might influence the patient’s quality of life, decrease their sensitivity to treatment, and finally shorten their survival rate [
3,
4]. According to the international consensus, three criteria can be used to diagnose cancer cachexia: a weight loss > 5% over the past six months, a body mass index < 20 and any degree of weight loss > 2%, or an appendicular skeletal muscle index consistent with sarcopenia and any degree of weight loss > 2%. The consensus also distinguishes three stages of cancer cachexia: precachexia, cachexia, and refractory cachexia [
1].
Research shows that the size of cancerous tissue is not associated with the presence of cachexia, but that the stage of cancer can affect the severity of cachexia. The prevalence of cancer cachexia is approximately 50% in all patients with cancer, increasing to 80% according to the disease progression [
5]. Almost 80% of patients with gastric or pancreatic cancer and 50% of patients with lung, prostate, or colon cancer experience cachexia. This syndrome also thrives in 40% of patients with breast tumors and some with leukemia [
6].
The pathogenesis of cancer cachexia is systemic inflammation caused by the interaction between tumor and organ tissues, resulting in alterations to the metabolism and homeostasis in different parts of the body [
5]. Cancer cachexia ultimately affects the skeletal muscle, but its effect is also found in other tissues, such as the cardiac muscle and liver [
7]. In the process of cancer cachexia, an imbalance between the synthesis and degradation of protein in the skeletal muscles’ activated signaling pathways potentially provokes muscle atrophy [
2]. Autophagy plays an important role in protein degradation, along with two other main pathways (the ubiquitin–proteasome system (UPS) and Ca
2+) [
2]. Autophagy is also an important regulator of the metabolism and homeostasis in many organs. Therefore, its involvement in skeletal muscle atrophy, cardiac remodeling, and the liver metabolism of cancer cachexia should be taken into consideration.
Studies have shown the complicated and changing role of autophagy in cancer [
8,
9,
10]. In the early stage of cancer, autophagy acts as a tumor suppressor; thus, increasing autophagy could prevent cancer initiation [
8]. However, in the advanced stages of cancer, autophagy is used by cancer cells to improve their fitness; therefore, inhibiting autophagy in cancer cells might serve as an option to improve cancer therapy [
10]. As such, understanding the phase of cancer is important for recognizing the importance of autophagy activity. For cancer cachexia, which more prominently occurs in the later stages of cancer, the disbalance of autophagy causes uncontrolled cytokine production and secretion by inflammatory cells in the tumor and other organs [
5]. Until recently, it has been debated whether using an autophagy modulator for cancer therapy is a wise decision, considering that autophagy also plays an important role in physiological functioning to maintain homeostasis [
10].
Exercise, which is also well-known to modulate autophagy, is one of the best treatment options to support the recovery of patients with cancer cachexia [
11]. The American College of Sports Medicine recommends 150 min (three to five days a week) of moderate-intensity aerobic exercise, two to three days a week of resistance exercise, and daily stretching for cancer survivors [
12]. Preclinical studies in rodents concluded that exercise does not affect cancer cachexia, but could reduce the severity of cachexia in the later stages of cancer [
13]. Human studies, on the other hand, have shown improvement of body weight and muscle mass in cachexia patients with lung and pancreatic cancer [
14]. A combined exercise program might be a better choice for improving muscle wasting in cancer cachexia [
15]. Recent studies have sought to delve deeper into the molecular mechanisms that occur in exercise before and after the development of cancer cachexia, especially those related to autophagy [
16,
17,
18,
19,
20,
21,
22,
23,
24]. Therefore, understanding autophagy modulation in the skeletal muscle, cardiac muscle, and liver after exercise in cancer cachexia is of the utmost importance for suggesting exercise as an autophagy modulator in different stages of cancer.
3. Changes of Autophagy in Organs Related to Cancer Cachexia
Autophagy is a physiological intracellular process that consists of the destruction and elimination of substances, such as misfolded proteins or organelles, to adapt or maintain cellular homeostasis. This process is mediated by an autophagosome, which comprises a double-layer vesicle and undergoes fusion of the autophagosome to the lysosome [
25]. Autophagy is a mandatory mechanism to maintain cell survival [
26,
27]. Many factors can induce autophagy, such as fasting, exercise, aging, disuse, and cancer [
28,
29,
30,
31,
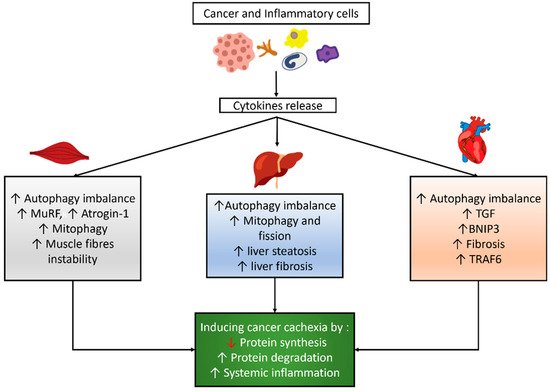
32]. Cancer and inflammatory cells might induce cytokine release, which influences the autophagy balance, mitophagy, and other signaling pathways in skeletal and cardiac muscles and the liver, as shown in
Figure 1.
Figure 1. Autophagy modulation and its correlated effects in cancer cachexia. Cancer and inflammatory cells induce cytokines release which then results in an imbalance of autophagy and other effects in skeletal muscle, cardiac muscle, and the liver, which eventually lead to cancer cachexia.
3.1. Autophagy Modulation in the Skeletal Muscle of Cancer Cachexia
Two main pathways induce protein degradation in cancer cachexia: UPS and autophagy [
2]. UPS requires enzymes called E1, E2, and E3 to be activated and docked to a protein that will be degraded. E3 binds to a specific protein that soon will be docked to ubiquitin and transported to the proteasome. Two kinds of E3 have been recognized to be involved in muscle atrophy: MuRF1 and Atrogin-1. The skeletal muscles in cancer cachexia also expressed MuRF1 and Atrogin-1, which activate E3 enzymes, thus stimulating UPS pathways [
33]. Autophagy also occurs through the process of engulfing ubiquitinated proteins via phagosomes and fusing them with a lysosome [
34].
Hentilä et al. reported higher levels of lipidated LC3, LC3II/LC3I ratio, LC3b mRNA, and Beclin-1 in the skeletal muscle of C26 tumor-bearing mice. These findings were followed by increased p62 protein and unchanged p62 mRNA levels, suggesting a reduction in autophagosome clearance. These findings imply that autophagy, in conjunction with the UPS, leads to muscle atrophy [
35]. Penna et al. measured the level of autophagy in the gastrocnemius muscle of C26 tumor-bearing mice and found that the levels of microtubule-associated protein 1 light chain 3B isoform I (LC3BI) remained unchanged, although the lipidated form (LC3BII) dramatically increased. Beclin-1, a key upstream regulator of autophagic sequestration, was significantly higher, implying that autophagy activation is a key to the early event in tumor-induced muscle depletion; p62 accumulated in a similar manner to Beclin-1 and LC3BII, which could indicate either the induction of autophagic sequestration or reduced autophagosome clearance [
32].
3.2. Autophagy Modulation in the Cardiac Muscle of Cancer Cachexia
In addition to skeletal muscle atrophy, cardiac remodeling is also induced by cancer cachexia, causing a condition known as cardiac cachexia [
34,
36]. Cancer cachexia might also induce cardiac remodeling that might disturb cardiac function, which is characterized by microscopic changes displaying cardiac atrophy because of the progressive loss of cardiac muscles [
36,
37].
The molecular mechanism underlying cardiac muscle atrophy is slightly different from the one that induces skeletal muscle atrophy in cancer cachexia [
34,
38]. Some studies showed an increase of UPS in the cachectic heart, although another study demonstrated that UPS did not increase, while autophagy did [
38,
39,
40,
41]. Cancer-induced cachexia implies a nutrient imbalance and cytokine production that affect cardiac muscle gene expression, causing an imbalance in cellular metabolism, inflammation, and necrosis [
42]. In such conditions, cardiac muscles express transforming growth factor (TGF) and BNIP3, which induce autophagy, and then necrosis and fibrosis development [
20]. Other markers, such as TRAF6, which is involved in apoptosis, and Beclin-1, which induces autophagy, are increased in cardiac muscle undergoing remodeling [
34].
Cosper and Leinwand proved that the proteins of cathepsin L, Beclin, and LC3 were increased during cardiomyocyte atrophy in cancer, suggesting increased autophagy. They found a two-fold increase of cathepsin L mRNA in both male and female atrophic hearts, a 1.5-fold increase of LC3 mRNA, and a seven-fold increase of LC3II protein levels in male atrophic hearts, but only a three-fold increase in females [
39]. Another study found a decrease of cardiac mass in the ApcMin/+ mouse model of colorectal cancer, together with an increase of Beclin-1 protein level, without a change of ubiquitination or apoptosis protein in the cardiac tissue. This result showed an increased autophagy process in cardiac cachexia, with no change of UPS stimulation and apoptosis [
43].
3.3. Autophagy Modulation in the Liver of Cancer Cachexia
Liver metabolism is also affected by cachexia via insufficient energy expenditure, phosphorylation oxidation, and lipid metabolism. The liver in cancer cachexia undergoes energy wasting by employing metabolic futile cycles that dissipate energy without anabolic or catabolic function [
44]. Tumor cells induce the liver to produce energy via gluconeogenesis of lactate that is derived. The lactate is produced by tumor cells through aerobic glycolysis, known as the “Warburg effect” [
45,
46]. The liver also releases acute-phase proteins that alleviate inflammation and increase muscle protein breakdown [
47,
48]. Amino acids produced by skeletal muscle protein degradation provide further gluconeogenesis in the liver [
44]. Energy wasting in cancer cachexia also decreases the phosphorylation oxidation capacity in mitochondria, which is marked by an increase of cardiolipin mediated by TNFα [
49,
50]. Recent research also showed that mitochondrial quality control (mitophagy and fission) was altered in cancer cachexia, in line with the development of hepatic fibrosis [
51]. The liver cells in patients with cachexia have a decreased capability to distribute fat via very low-density lipoprotein (VLDL), along with an inability to oxidize fat production and deposition in liver cells, thus leading to liver steatosis [
48]. These changes in liver metabolism are correlated with autophagy function, especially lipid droplet clearance, damaged mitochondria, protein aggregate removal, and liver fibrosis prevention [
52]. Previous research strongly demonstrated that autophagy is critical to liver function, with lower autophagy linked to poorer results [
53,
54,
55].
Rosa-Caldwell et al. showed that there was no link between the Beclin mRNA content and cancer progression. It was also observed that the LC3II/LC3I ratio, which acted as a surrogate measure of autophagosome production, showed a quadratic connection with cancer progression, with a 40–50% decrease in the LC3II/LC3I ratio at weeks one, two, and three, subsequently increasing at week four. The level of p62 mRNA did not change between the cancer groups, whereas the level of p62 protein increased over time as the disease progressed. In addition, it was also found that mitophagy and fission are altered before hepatic fibrosis occurs [
51]. Another study found that the level of LC3II and the LC3II/LC3I ratio, as well as Beclin-1, increased in the liver of C26 tumor-bearing animals. In contrast to the skeletal muscle, an animal model of C26 colon cancer only showed elevated LC3I in the liver, with no change in p62, suggesting increased autophagosome formation without increased or decreased autophagic flux [
35].
This entry is adapted from the peer-reviewed paper 10.3390/life11080781