Sphingolipids are both structural molecules that are essential for cell architecture and second messengers that are involved in numerous cell functions. Ceramide is the central hub of sphingolipid metabolism. In addition to being the precursor of complex sphingolipids, ceramides induce cell cycle arrest and promote cell death and inflammation. At least some of the enzymes involved in the regulation of sphingolipid metabolism are altered in carcinogenesis, and some are targets for anticancer drugs.
- ceramide (Cer)
- sphingolipids (Sphs)
- cancer
- ceramide 1-phosphate (C1P)
- shingosine 1-phosphate (S1P)
- deoxy-sphingolipids
- apoptosis
- cell proliferation
1. Introduction
Sphingolipids are fundamental components of cell membranes. They were discovered in brain extracts by J. L. W. Thudichum in 1876 and were considered merely as structural molecules of cell architecture. It was not until 1986 when Hannun and co-workers demonstrated that some sphingolipids were bioactive molecules, showing that sphingosine was capable of inhibiting protein kinase C (PKC) [1]. Since then, many sphingolipids have been shown to play critical roles in cell activation and the control of cell and tissue homeostasis [2][3][4]. Among all of the sphingolipids that are present in the organism, sphingosine and ceramide, together with their phosphorylated forms sphingosine-1-phosphate (S1P) and ceramide-1-phosphate (C1P), are crucial regulators of cell physiology and pathology [5][6].
So far, more than 30 enzymes involved in sphingolipid metabolism have been characterized. These enzymes are highly regulated and are involved in the regulation of relevant pathophysiologic processes. Dysfunction of these enzymes may cause decompensation of the concentrations of their sphingolipid products, leading to a loss of cellular homeostasis. An important aspect that demonstrates the specificity and importance of sphingolipids in cell physiology is that small variations in sphingolipid concentrations can lead to or be a consequence of disease [4]. Among the most important sphingolipids are ceramide (Cer), sphingosine (Sph) and their phosphorylated forms, C1P and S1P. The intracellular concentrations of these metabolites define a precise balance that eventually determines cellular actions and fate. Whilst Cer and Sph are proapoptotic, C1P and S1P are proliferative and anti-apoptotic signals [4]. Sph is the precursor of S1P through phosphorylation by sphingosine kinases 1 and 2 (SphK1 and SphK2) [7][8], whereas C1P is produced by the action of CerK. Whether CerK exists as a sole gene product or as a family of different isoforms awaits further investigation. CerK resides in the Golgi apparatus, where it can phosphorylate ceramide that is transported by ceramide transfer protein (CERT) from the ER to the Golgi. Noteworthily, CerK-generated C1P can be transported by a recently identified C1P transfer protein (CPTP) from the Golgi to the plasma membrane, where it might participate in signal transduction processes [9]. Although CerK has also been identified in plants [10], it is the only enzyme capable of generating C1P in mammalian cells [11]. As mentioned above, an appropriate equilibrium between sphingosine and ceramide, versus their phosphorylated forms, can determine cell function and fate (Figure 1). When this balance is disturbed or altered, disease may arise. In particular, many inflammatory processes and pathologies, including cardiovascular diseases, neurodegenerative disorders, lung inflammatory disorders (such as chronic obstructive pulmonary disease (COPD) or asthma) type 2 diabetes or cancer, are characterized by disruption of sphingolipid homeostasis [12][13][14][15][16][17][18][19][20][21]. Moreover, it has been reported that treatment with short-chain Cer anologs is an effective treatment against certain cancers [22][23][24]. In addition, deoxy-sphingolipids have been shown to be of great importance when observing their potential as regulators of sphingolipid metabolism.
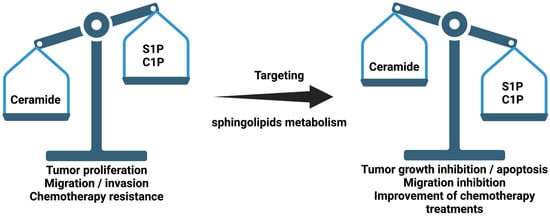
Figure 1. Schematic representation of sphingolipid ratio and their relationship with cancer. Sphingosine 1-phosphate (S1P) and ceramide 1-phosphate (C1P).
2. Enzymes of Ceramide Metabolism
2.1. The De Novo Pathway of Ceramide Synthesis
2.1.1. Serine Palmitoyl Transferase (SPT)
Serine palmitoyl transferase (SPT) is a heteromeric enzyme, composed of three subunits, that catalyzes the first step of the de novo ceramide synthesis pathway. The protein is localized to the endoplasmic reticulum. It catalyzes the condensation of L-serine and palmitoyl-coenzyme A to generate 3-keto-sphinganine (Figure 1). Several studies have described an increase in SPT activity in response to chemotherapy and radiotherapy in various types of cancer. So far, some SPT inhibitors that block tumor growth have been described. In particular, myriocin (also known as ISP-1) is a potent SPT inhibitor [25]. It was discovered as an antifungal antibiotic from Myriococcum albomyces [26], and later it was shown to block SPT [27]. Treatment with myriocin caused growth inhibition of breast cancer [19] and B16F10 melanoma cells through G2/M phase arrest [28]. In addition, there is a positive correlation between SPT enzyme inhibition and growth suppression of a human lung adenocarcinoma cell line (HCC4006) [29]. Furthermore, inhibition of SPT with myriocin or knocking down the enzyme expression with a specific siRNA inhibited the proliferation of human U87MG glioblastoma cells, an action that was associated with suppression of intracellular S1P levels [30]. Synthetic molecules of imidazopyridine and pyrazolopiperidine derivatives also showed high affinity and inhibitory capacity for SPT, decreasing ceramides levels in serum by 50% in mice that were treated orally for one week, and they were proposed as potential treatments for diabetes [31]. The authors of the latter report also designed two new inhibitors of SPT, tetrahydropyrazolopyridine and 3-phenylpiperidine. Both of these compounds were able to reduce ceramide concentration in the blood, and they inhibited the proliferation of HCC4006 and PL-21 cells derived from an acute promyelocytic leukemia [32][33].
It has been proposed that the antitumor activity of SPT is caused by increasing the levels of proapoptotic ceramides. In fact, some anticancer drugs exert at least part of their therapeutic effects through the activation of SPT. In particular, fenretinide, a synthetic retinoid and cytotoxic molecule to a variety of cancer cells, increased the concentration of dhCer and Cer, leading to neuroblastoma cell apoptosis, an action that was associated with the interaction of this drug with SPT and DeS1 [34]. Furthermore, a fenretinide metabolite, 4-oxo-fenretinide, markedly induced G2-M cell cycle arrest and apoptosis in ovarian, breast and neuroblastoma tumor cells [35]. Interestingly, naturally occurring resveratrol, a phytoalexin present in grapes and red wine, potently induced growth inhibition and apoptosis in metastatic breast cancer cells via the activation of SPT and accumulation of ceramide [36].
Recently, it has been shown that the SPTLC2 and SPTLC3 subunits have a different affinity for Acyl-CoA in the function of the chain, giving rise to Cer of different lengths, while SPTLC1 is essential to produce an active enzyme [37][38]. ssSPT subunits have been described as important regulators of SPT activity. Recently, it has been observed that in patients with prostate cancer treated with anti-androgens treatment, an increase in the expression of SPTSSB was detected [39]. Moreover, administration of exogenous Cer nanoliposomes was determined as an efficient treatment in cancer cells [39]. In addition to ssSPT subunits, the Tsc3 subunit was found to be a potent regulatory protein of SPT, increasing its activity by up to 50-fold [40][41].
2.1.2. Ceramide Synthase (CerS)
CerS takes part in both the de novo pathway and the salvage pathway of ceramide synthesis. CerS is a family of six different isoforms, each of which synthesizes ceramides with different chain lengths of fatty acyl-CoA, resulting in a specific activity. This fact is important, since, for example, CerS1 generates 18-carbon Cer (C18:0-Ceramide) that inhibits tumor growth [42], whereas CerS5/6 produces C16:0-Ceramide, an anti-apoptotic metabolite in human head and neck squamous cell carcinomas [43] that is also involved in type II diabetes [17][44][45] and that has proinflammatory properties [46]. Interestingly, a specific inhibitor of CerS1, called P053, has been described to reduce the levels of C18:0-Ceramide in renal HEK 293 cancer cells [47]. Overexpression of CerS2 or CerS4 increases very long chain ceramides (C24:0-Ceramide), which have a protective effect on human breast and colon cancer [48]. Recently, increased levels of C24:0-Ceramide in gallbladder cancer was observed due to the overexpression of SPT and CerS2 and related to the progression of the tumor [24].
Interestingly, analogs derived from Fingolimod (FTY720) specifically inhibit isoforms of CerS in HCT-106 and HeLa cells. Specifically, the ST1058 and ST1074 inhibitors preferentially inhibit CerS2 and CerS4, whereas ST1072 blocks the activities of CerS4 and CerS6, and ST1060 inhibits CerS2 [49].
It has also been shown that overexpression of CerS in certain types of cancer confers resistance to chemotherapy [50]. Concerning anticancer drugs, anthracyclines have been shown to increase the content of pro-apoptotic ceramides by stimulating CerS [51]. In particular, vinblastine activated CerS, increasing ceramide content in the liver [52]; paclitaxel (taxol) is an antitumor molecule that led to Cer-induced apoptosis in human breast cancer cells by activation of both SPT and CerS [53], and antifolate methotrexate (MTX) increased the levels of C16:0-Ceramide to reduce proliferation in a p53-dependet manner in human lung adenocarcinoma cells [54]. In addition, a recent study showed that the activity of CerS6, giving rise to Cer, increases the inhibitory capacity of p53 to block progeny formation in polyploid cancer cells [55]. By contrast, fumonisin B1, a fungal metabolite that potently inhibits CerS, was shown to reduce apoptotic cell death in a variety of cell types, including SCC17B human head and neck squamous carcinoma cells [56].
2.1.3. Dihydroceramide Desaturase (Des1, also Known as DEGS1)
Des1 is the last enzyme in the de novo synthesis pathway of ceramide. This enzyme catalyzes the conversion of dhCer to Cer by insertion of a trans double bond in position 4–5. Experiments using heterozygous DES1-null mice showed higher dhCer/Cer ratios in multiple organs, rendering cells resistant to apoptosis [57]. Furthermore, knockdown of Des1 using siRNA [58] led to cell cycle arrest in human neuroblastoma cells [58]. However, accumulation of dhCer in cells can stimulate autophagy. Although autophagy is a mechanism of cancer cell survival, it can also lead to cell death [59][60][61]. So far, several compounds with the ability to inhibit Des1 have been described. For example, resveratrol, which is a dietary polyphenol with well recognized antioxidant and health beneficial properties, was shown to inhibit Des1, causing autophagy in gastric cancer HGC27 cells [62]. Other Des1 inhibitors, including γ-tocotrienol, phenoxodiol or celecoxib, induce autophagy by dhCer accumulation in T98G and U87MG glioblastoma cell lines by inhibition of Des1 [59]. Furthermore, 3-ketosphinganine and its dideuterated analog at C4 (d2KSa) are metabolized to produce high levels of dihydrosphingolipids (dhSLs) in HGC27, T98G and U87MG cancer cells. d2KSa provokes the accumulation of dhCer and other dhSLs by inhibition of Des1 and induces autophagy in cancer cells [63]. Fenretinide, which is an agonist of SPT, also inhibits Des1 and, combined with Foscan-mediated photodynamic therapy, enhances apoptosis in SCC19 cells, a human head and neck squamous cell carcinoma [64].
N-[(1R,2Shttpo)-2-hydroxy-1-hydroxymethyl-2-(2-tridecyl-1-cyclopropenyl)ethyl]octanamide (GT11) is a specific inhibitor of Des1 that efficiently activates autophagy and apoptosis of the human U87MG glioma cell line. In addition, treatment with tetrahydrocannabinol (THC) produces an alteration of the lipid composition in the endoplasmic reticulum and, along with it, the accumulation of dhCer due to the reduction of Des1 expression, leading to a stimulation of autophagy and apoptosis in human U87MG glioma cells [60]. Moreover, THC treatment or selective inhibition of Des1 by GT11 efficiently activates autophagy and apoptosis and inhibits tumor growth in mice [60].
2.2. Enzymes of the Sphingomyelinase Pathway
2.2.1. Sphingomyelinases (SMase)
Based on their optimal pH for activity and their location, there are three major types of SMases: aSMase, nSMase and Alk-SMase. SMases catalyze the breakdown of membrane SM to ceramide and phosphocholine. Stimulation of SMase activity is related to oxidative stress [65], cell death signals such as TNF Receptor (TNFR) and Fas [66][67] or anticancer drugs [68]. Experiments where SMase activity was knocked down showed inhibition of tumor growth in non-small cell lung carcinomas [69]. Furthermore, aSMase-null mice (smpd1-/-) were protected from pulmonary tumor metastasis [70]. Contrarily, resveratrol was described to inhibit cell proliferation of human leukemia (K562 cell line) and cancer cells (HCT116 cell line) by up-regulation of aSMase and the consequent Cer accumulation [71].
The term FIASMA (Functional Inhibitor of Acid SphingoMyelinAse) encompasses a family of drugs, many of them approved for use in humans, which have been shown to indirectly inhibit the activity of aSMase [72]. Interestingly, recent studies have shown that inhibition of lysosomal SMase via these inhibitors prevents infection of the SARS-CoV-2 virus [73][74][75]. In Niemann–Pick disease, it has been observed that aSMase dysfunction leads to a dysregulation of lysosomes, causing a disruption of cholesterol transport [76]. This process can lead to cell dysfunction and death. Perphenazine and fluphenazine, both FIASMA members, were found to inhibit xenografted tumor growth by disruption of intracellular cholesterol transport [77].
Recently, a small molecule called ARC39 was synthetized and characterized. ARC39 was found to inhibit both lysosomal and secretory aSMase in vitro with L929, HepG2 and B16F10 cells [78]. However, ARC39 inhibition in vivo could not be confirmed; therefore, more studies should be carried out. Moreover, a potent light-inducible photocaged ASM inhibitor (PCAI) with the ability to inhibit aSMase has been developed. Furthermore, being inducible by light, it is addressable [79].
Most studies on the implication of SMase activity in cancer are related to the acidic form of the enzyme, although nSMase may also play an important role in cancer biology [67]. nSMase has also been observed to be involved in the activation and migration of T cells, responsible for the detection and elimination of cancer cells [80]. Doxorubicin is an antibiotic of the anthracycline family widely used in chemotherapy treatment. Interestingly, doxorubicin-induced growth arrest in breast cancer cells is due to a p53-dependent upregulation of nSMase-2 [68]. As seen above, chemotherapy treatments are highly linked to the metabolism of sphingolipids, sometimes in a good way and others in the opposite direction. Recently, it has been observed that doxorubicin treatment stimulated the activity of aSMase, leading to an increase in Cer and reactive oxygen species (ROS), promoting apoptosis [81]. As a side effect, the release of ROS into the bloodstream damaged the vascular endothelium [81]. Exosomes are vesicles between 40–160 nm that can be secreted by a variety of cells. They carry nucleic acids, proteins and metabolites. Their function is that of intercellular communication between nearby and distant cells, both in physiological conditions and in certain diseases. In recent years, their involvement as a biomarker and therapeutic target has gained great interest [82][83]. Exosomes have been found to be crucial for the formation and maintenance of microenvironments in tumors. The need for nSMase activity for the release of exosomes has been described [82][84]. Therefore, certain studies in tumors have focused on the inhibition of this enzyme. In particular, the nSMase isoform, nSMase2, was involved in exosome miRNA secretion and contributed to cancer cell metastasis through the induction of angiogenesis [85]. Specific inhibition of nSMase by GW4869 showed a potential target to block the metastasis. In addition, low expression of nSMase was found in exosomes from hepatocellular carcinoma patients, related with a poor outcome [86]. Similarly, exosomes have been shown to be signaling systems for the immune system, allowing the fight against tumors. Recently, it has been observed that the treatment with platinium nanoparticles enhanced exosomes secretion in A549 lung cancer cells [87].
2.2.2. Sphingomyelin Synthase (SMS)
The sphingomyelin synthase (SMS) family consists of three members, SMS1, SMS2 and SMS-related protein (SMSr) [88]. This enzyme activity catalyzes the synthesis of SM from Cer and phosphatidylcholine, thereby releasing diacylglicerol. It was demonstrated that selective inhibition of SMS by D609 induces an increase in the concentration of ceramide in the ER, and it triggers autophagy in hippocampal neurons [89]. However, in glioblastoma, it was observed that treatment with the 2-hydroxyoleic acid (2OHOA), an anticancer drug, caused an increase in SMS activity. Furthermore, the inhibition with D609 diminishes the anticancer effect [90]. Recently, SMS2 was found to be upregulated in different cancers, such as breast [91] and ovarian [92]. High infiltration of M2-polarized macrophages in primary tumors is related with poor outcomes in patients. Zheng and co-workers demonstrated the disruption of apoptotic pathways by activation of SMS2 in breast cancer [93]. Activation of SMS2 causes a reduction in Cer levels and stimulates cell proliferation through activation of the transforming growth factor-β (TGF-β)/Smad signaling pathway. By contrast, inhibition of SMS2 by specific siRNA led to accumulation of Cer and the promotion of cell death [93]. In addition, recent works observed that SMS2 is upregulated in breast cancer and induces macrophages polarization and tumor progression [91]. Moreover, SMS2 inhibition by 15w or knockdown reduced the release of cytokines that stimulate macrophage polarization, reducing tumor growth [91]. In addition, SMS1 down regulation was found in metastatic melanoma patients and was associated with worse prognosis due to an imbalance between SM and glucosyl-ceramide levels [94].
2.3. Enzymes of the Salvage Pathway
2.3.1. Glucosylceramide Synthase (GCS)
Glucosylceramide synthase (GCS) is a lysosomal enzyme that introduces a glycosylation on the ceramide to produce glucosylceramide. GCS has been observed to be upregulated in different types of cancer, being associated with resistance to anticancer treatments [95][96][97][98][99]. Glucosylceramide plasma levels have recently been described as a promised biomarker for prostate cancer status [100]. A recent study has demonstrated that a GCS inhibitor, called 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP), caused a lysosomal lipid accumulation and inactivation of mTOR in HeLa cells, leading to apoptotic stimulation [101].
2.3.2. Acid β-Glucosidase (β-GCase)
Mutations in the GBA1 gene that codes for β-GCase is one of the main causes of the development of Parkinson’s disease [102][103]. Different studies have associated the deficiency of this gene with the possibility of suffering from certain types of cancer [104][105]. Recently, it was reported that β-GCase was overexpressed in chemotherapy-resistant gastric cancer cells [106]. In addition, it was observed that its inhibition with specific siRNAs produced a lysosomal dysfunction that, together with anticancer treatment, caused cell death [106].
2.3.3. Ceramidase
As seen above, ceramidases catalyze the degradation of ceramide to sphingosine. Ceramidases have been described as important regulators of the processes of cellular autophagy and resistance to chemotherapy. Therefore, inhibition of the different ceramidase isoforms has been shown to be beneficial for the treatment of different types of cancer [107][108][109]. In recent years, different inhibitors have been developed that have shown their potential as an anticancer therapy [110].
Acid ceramidase (ASAH1) has been detected to be overexpressed in melanomas, conferring resistance to chemotherapy. The involvement of ASAH1 in mitochondrial function and cellular autophagy in melanoma cells has been observed [111]. Furthermore, a recent study has shown that deletion of the ASAH1 gene blocks the induction of apoptosis mediated by doxorubicin [112]. These results may be due to the high adaptive capacity of melanoma cells. However, inhibition of ASAH1 with siRNA or pharmacologically with LCL521 in colorectal cancer cell lines (HT29, HCT116 and LIM1215) enhanced X-ray radiosensitivity [113]. Moreover, tamoxifen has shown its ability to inhibit ASAH1 in polyploid giant cancer cells by reducing their division [114]. Recently, the synthesis of N-metallocenoylsphingosines has been described, demonstrating its cytotoxic capacity and inhibition of the cell cycle in cancer cells [115].
Alkaline ceramidase (ACER) has been described as being highly related to the growth, migration and invasion of tumor cells [116][117][118]. In this sense, incubation of HepG2 and Huh-7 human liver cancer cells with the ACER2 inhibitor, called D-erythro-MAPP (D-e-MAPP), has been shown to reduce cell growth [116]. Structural analogues of ceramides have demonstrated their efficacy as selective ACER inhibitors, blocking cell growth and being suitable candidates for anticancer treatment [119][120]. More specifically, deoxy-sphingolipid analogs have recently been described as highly selective molecules and potent ACER3 inhibitors, with the ability to inhibit the cell cycle [121]. Of interest, the structure of ACER3 has recently been described, allowing structural and computational studies for the development of new specific inhibitors [122].
This entry is adapted from the peer-reviewed paper 10.3390/medicina57070729
References
- Hannun, Y.A.; Loomis, C.R.; Merrill, A.H., Jr.; Bell, R.M. Sphingosine Inhibition of Protein Kinase C Activity and of Phorbol Dibutyrate Binding in Vitro and in Human Platelets. J. Biol. Chem. 1986, 261, 12604–12609.
- Hannun, Y.A.; Obeid, L.M. Principles of Bioactive Lipid Signalling: Lessons from Sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150.
- Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabrias, G.; Abad, J.L.; Delgado, A.; et al. Control of Metabolism and Signaling of Simple Bioactive Sphingolipids: Implications in Disease. Prog. Lipid Res. 2010, 49, 316–334.
- Gomez-Larrauri, A.; Presa, N.; Dominguez-Herrera, A.; Ouro, A.; Trueba, M.; Gomez-Munoz, A. Role of Bioactive Sphingolipids in Physiology and Pathology. Essays Biochem. 2020, 64, 579–589.
- Arana, L.; Gangoiti, P.; Ouro, A.; Trueba, M.; Gomez-Munoz, A.; Gómez-Muñoz, A. Ceramide and Ceramide 1-Phosphate in Health and Disease. Lipids Health Dis. 2010, 9, 15.
- Gangoiti, P.; Granado, M.H.; Wei, S.; Kong, J.Y.; Steinbrecher, U.P.; Gómez-muñoz, A. Ceramide 1-Phosphate Stimulates Macrophage Proliferation through Activation of the PI3-Kinase/PKB, JNK and ERK1/2 Pathways. Cell. Signal. 2008, 20, 726–736.
- Maceyka, M.; Payne, S.G.; Milstien, S.; Spiegel, S. Sphingosine Kinase, Sphingosine-1-Phosphate, and Apoptosis. Biochim. Biophys. Acta 2002, 1585, 193–201.
- Taha, T.A.; Hannun, Y.A.; Obeid, L.M. Sphingosine Kinase: Biochemical and Cellular Regulation and Role in Disease. J. Biochem. Mol. Biol. 2006, 39, 113–131.
- Simanshu, D.K.; Kamlekar, R.K.; Wijesinghe, D.S.; Zou, X.; Zhai, X.; Mishra, S.K.; Molotkovsky, J.G.; Malinina, L.; Hinchcliffe, E.H.; Chalfant, C.E.; et al. Non-Vesicular Trafficking by a Ceramide-1-Phosphate Transfer Protein Regulates Eicosanoids. Nature 2013, 500, 463–467.
- Liang, H.; Yao, N.; Song, J.T.; Luo, S.; Lu, H.; Greenberg, J.T. Ceramides Modulate Programmed Cell Death in Plants. Genes Dev. 2003, 17, 2636–2641.
- Boath, A.; Graf, C.; Lidome, E.; Ullrich, T.; Nussbaumer, P.; Bornancin, F. Regulation and Traffic of Ceramide 1-Phosphate Produced by Ceramide Kinase: Comparative Analysis to Glucosylceramide and Sphingomyelin. J. Biol. Chem. 2008, 283, 8517–8526.
- Guan, X.L.; Mäser, P. Comparative Sphingolipidomics of Disease-Causing Trypanosomatids Reveal Unique Lifecycle- and Taxonomy-Specific Lipid Chemistries. Sci. Rep. 2017, 7, 1–13.
- Mielke, M.M.; Haughey, N.J.; Bandaru, V.V.R.; Zetterberg, H.; Blennow, K.; Andreasson, U.; Johnson, S.C.; Gleason, C.E.; Blazel, H.M.; Puglielli, L.; et al. Cerebrospinal Fluid Sphingolipids, β-Amyloid, and Tau in Adults at Risk for Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 2486–2494.
- van Kruining, D.; Luo, Q.; van Echten-Deckert, G.; Mielke, M.M.; Bowman, A.; Ellis, S.; Oliveira, T.G.; Martinez-Martinez, P. Sphingolipids as Prognostic Biomarkers of Neurodegeneration, Neuroinflammation, and Psychiatric Diseases and Their Emerging Role in Lipidomic Investigation Methods. Adv. Drug Deliv. Rev. 2020, 159, 232–244.
- Griese, M.; Bonella, F.; Costabel, U.; de Blic, J.; Tran, N.-B.; Liebisch, G. Quantitative Lipidomics in Pulmonary Alveolar Proteinosis. Am. J. Respir. Crit. Care Med. 2019, 200, 881–887.
- Kurek, K.; Mikłosz, A.; Łukaszuk, B.; Chabowski, A.; Górski, J.; Zendzian-Piotrowska, M. Inhibition of Ceramide de Novo Synthesis Ameliorates Diet Induced Skeletal Muscles Insulin Resistance. J. Diabetes Res. 2015, 2015.
- Li, C.; Zhou, J.; Wang, A.; Li, N.; Chen, B.; Lei, M. Inhibition of Ceramide Synthesis Reverses Endothelial Dysfunction and Atherosclerosis in Streptozotocin-Induced Diabetic Rats. Diabetes Res. Clin. Pract. 2011, 93, 77–85.
- Alberg, A.J.; Armeson, K.; Pierce, J.S.; Bielawski, J.; Bielawska, A.; Visvanathan, K.; Hill, E.G.; Ogretmen, B. Plasma Sphingolipids and Lung Cancer: A Population-Based, Nested Case-Control Study. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1374–1382.
- Ogretmen, B. Sphingolipid Metabolism in Cancer Signalling and Therapy. Nat. Rev. Cancer 2018, 18, 33–50.
- Custodia, A.; Aramburu-Núñez, M.; Correa-Paz, C.; Posado-Fernández, A.; Gómez-Larrauri, A.; Castillo, J.; Gómez-Muñoz, A.; Sobrino, T.; Ouro, A. Ceramide Metabolism and Parkinson’s Disease-Therapeutic Targets. Biomolecules 2021, 11, 945.
- Sheridan, M.; Ogretmen, B. The Role of Ceramide Metabolism and Signaling in the Regulation of Mitophagy and Cancer Therapy. Cancers 2021, 13, 2475.
- Wilhelm, R.; Eckes, T.; Imre, G.; Kippenberger, S.; Meissner, M.; Thomas, D.; Trautmann, S.; Merlio, J.P.; Chevret, E.; Kaufmann, R.; et al. C6 Ceramide (D18: 1/6: 0) as a Novel Treatment of Cutaneous T Cell Lymphoma. Cancers 2021, 13, 270.
- Xu, Y.; Pan, J.; Lin, Y.; Wu, Y.; Li, H.; Chen, Y. C6-Ceramide Induces Apoptosis in Lung Non-Small Cell Lung Cancer and Suppresses Brain Metastasis by Downregulating the PI3K/AKT/MTOR Signaling Pathway. Res. Sq. 2021.
- Zhang, Y.; Wang, H.; Chen, T.; Wang, H.; Liang, X.; Zhang, Y.; Duan, J.; Qian, S.; Qiao, K.; Zhang, L.; et al. C24-Ceramide Drives Gallbladder Cancer Progression Through Directly Targeting Phosphatidylinositol 5-Phosphate 4-Kinase Type-2 Gamma to Facilitate Mammalian Target of Rapamycin Signaling Activation. Hepatology 2021, 73, 692–712.
- Glaros, E.N.; Kim, W.S.; Wu, B.J.; Suarna, C.; Quinn, C.M.; Rye, K.A.; Stocker, R.; Jessup, W.; Garner, B. Inhibition of Atherosclerosis by the Serine Palmitoyl Transferase Inhibitor Myriocin Is Associated with Reduced Plasma Glycosphingolipid Concentration. Biochem. Pharmacol. 2007, 73, 1340–1346.
- Kluepfel, D.; Bagli, J.; Baker, H.; Charest, M.P.; Kudelski, A.; Sehgal, S.N.; Vézina, C. Myriocin, a New Antifungal Antibiotic from Myriococcum Albomyces. J. Antibiot. 1972, 25, 109–115.
- Riley, R.T.; Norred, W.P.; Wang, E.; Merrill, A.H. Alteration in Sphingolipid Metabolism: Bioassays for Fumonisin- and ISP-I-like Activity in Tissues, Cells and Other Matrices. Nat. Toxins 1999, 7, 407–414.
- Lee, Y.S.; Choi, K.M.; Choi, M.H.; Ji, S.Y.; Lee, S.; Sin, D.M.; Oh, K.W.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; et al. Serine Palmitoyltransferase Inhibitor Myriocin Induces Growth Inhibition of B16F10 Melanoma Cells through G2/M Phase Arrest. Cell Prolif. 2011, 44, 320–329.
- Sano, O.; Kazetani, K.; Adachi, R.; Kurasawa, O.; Kawamoto, T.; Iwata, H. Using a Biologically Annotated Library to Analyze the Anticancer Mechanism of Serine Palmitoyl Transferase (SPT) Inhibitors. FEBS Open Bio 2017, 7, 495–503.
- Bernhart, E.; Damm, S.; Wintersperger, A.; Nusshold, C.; Brunner, A.M.; Plastira, I.; Rechberger, G.; Reicher, H.; Wadsack, C.; Zimmer, A.; et al. Interference with Distinct Steps of Sphingolipid Synthesis and Signaling Attenuates Proliferation of U87MG Glioma Cells. Biochem. Pharmacol. 2015, 96, 119–130.
- Genin, M.J.; Gonzalez Valcarcel, I.C.; Holloway, W.G.; Lamar, J.; Mosior, M.; Hawkins, E.; Estridge, T.; Weidner, J.; Seng, T.; Yurek, D.; et al. Imidazopyridine and Pyrazolopiperidine Derivatives as Novel Inhibitors of Serine Palmitoyl Transferase. J. Med. Chem. 2016, 59, 5904–5910.
- Kojima, T.; Asano, Y.; Kurasawa, O.; Hirata, Y.; Iwamura, N.; Wong, T.T.; Saito, B.; Tanaka, Y.; Arai, R.; Yonemori, K.; et al. Discovery of Novel Serine Palmitoyltransferase Inhibitors as Cancer Therapeutic Agents. Bioorg. Med. Chem. 2018, 26, 2452–2465.
- Yaguchi, M.; Shibata, S.; Satomi, Y.; Hirayama, M.; Adachi, R.; Asano, Y.; Kojima, T.; Hirata, Y.; Mizutani, A.; Kiba, A.; et al. Antitumor Activity of a Novel and Orally Available Inhibitor of Serine Palmitoyltransferase. Biochem. Biophys. Res. Commun. 2017, 484, 493–500.
- Maurer, B.J.; Metelitsa, L.S.; Seeger, R.C.; Cabot, M.C.; Patrick Reynolds, C. Increase of Ceramide and Induction of Mixed Apoptosis/Necrosis by N-(4- Hydroxyphenyl)-Retinamide in Neuroblastoma Cell Lines. J. Natl. Cancer Inst. 1999, 91, 1138–1146.
- Villani, M.G.; Appierto, V.; Cavadini, E.; Bettiga, A.; Prinetti, A.; Clagett-Dame, M.; Curley, R.W.; Formelli, F. 4-Oxo-Fenretinide, a Recently Identified Fenretinide Metabolite, Induces Marked G2-M Cell Cycle Arrest and Apoptosis in Fenretinide-Sensitive and Fenretinide-Resistant Cell Lines. Cancer Res. 2006, 66, 3238–3247.
- Scarlatti, F.; Sala, G.; Somenzi, G.; Signorelli, P.; Sacchi, N.; Ghidoni, R. Resveratrol Induces Growth Inhibition and Apoptosis in Metastatic Breast Cancer Cells via de Novo Ceramide Signaling. FASEB J. 2003, 17, 2339–2341.
- Hornemann, T.; Penno, A.; Rütti, M.F.; Ernst, D.; Kivrak-Pfiffner, F.; Rohrer, L.; von Eckardstein, A. The SPTLC3 Subunit of Serine Palmitoyltransferase Generates Short Chain Sphingoid Bases. J. Biol. Chem. 2009, 284, 26322–26330.
- Lone, M.A.; Hülsmeier, A.J.; Saied, E.M.; Karsai, G.; Arenz, C.; von Eckardstein, A.; Hornemann, T. Subunit Composition of the Mammalian Serine-Palmitoyltransferase Defines the Spectrum of Straight and Methyl-Branched Long-Chain Bases. Proc. Natl. Acad. Sci. USA 2020, 117, 15591–15598.
- Costa-Pinheiro, P.; Heher, A.; Raymond, M.H.; Jividen, K.; Shaw, J.J.; Paschal, B.M.; Walker, S.J.; Fox, T.E.; Kester, M. Role of SPTSSB-Regulated de Novo Sphingolipid Synthesis in Prostate Cancer Depends on Androgen Receptor Signaling. IScience 2020, 23, 101855.
- Gable, K.; Slife, H.; Bacikova, D.; Monaghan, E.; Dunn, T.M. Tsc3p Is an 80-Amino Acid Protein Associated with Serine Palmitoyltransferase and Required for Optimal Enzyme Activity. J. Biol. Chem. 2000, 275, 7597–7603.
- Ren, J.; Saied, E.M.; Zhong, A.; Snider, J.; Ruiz, C.; Arenz, C.; Obeid, L.M.; Girnun, G.D.; Hannun, Y.A. Tsc3 Regulates SPT Amino Acid Choice in Saccharomyces Cerevisiae by Promoting Alanine in the Sphingolipid Pathway. J. Lipid Res. 2018, 59, 2126–2139.
- Wang, Z.; Wen, L.; Zhu, F.; Wang, Y.; Xie, Q.; Chen, Z.; Li, Y. Overexpression of Ceramide Synthase 1 Increases C18-Ceramide and Leads to Lethal Autophagy in Human Glioma. Oncotarget 2017, 8, 104022–104036.
- Moro, K.; Nagahashi, M.; Gabriel, E.; Takabe, K.; Wakai, T. Clinical Application of Ceramide in Cancer Treatment. Breast Cancer 2019, 26, 407–415.
- Chaurasia, B.; Tippetts, T.S.; Monibas, R.M.; Liu, J.; Li, Y.; Wang, L.L.; Wilkerson, J.L.; Rufus Sweeney, C.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a Ceramide Double Bond Improves Insulin Resistance and Hepatic Steatosis. Science 2019, 365, 386–392.
- Xia, Q.S.; Lu, F.E.; Wu, F.; Huang, Z.Y.; Dong, H.; Xu, L.J.; Gong, J. New Role for Ceramide in Hypoxia and Insulin Resistance. World J. Gastroenterol. 2020, 26, 2177–2186.
- Crivelli, S.M.; Luo, Q.; Stevens, J.A.A.; Giovagnoni, C.; van Kruining, D.; Bode, G.; den Hoedt, S.; Hobo, B.; Scheithauer, A.-L.; Walter, J.; et al. CERTL Reduces C16 Ceramide, Amyloid-β Levels, and Inflammation in a Model of Alzheimer’s Disease. Alzheimer’s Res. Ther. 2021, 13, 45.
- Turner, N.; Lim, X.Y.; Toop, H.D.; Osborne, B.; Brandon, A.E.; Taylor, E.N.; Fiveash, C.E.; Govindaraju, H.; Teo, J.D.; McEwen, H.P.; et al. A Selective Inhibitor of Ceramide Synthase 1 Reveals a Novel Role in Fat Metabolism. Nat. Commun. 2018, 9, 1–14.
- Hartmann, D.; Lucks, J.; Fuchs, S.; Schiffmann, S.; Schreiber, Y.; Ferreirós, N.; Merkens, J.; Marschalek, R.; Geisslinger, G.; Grösch, S. Long Chain Ceramides and Very Long Chain Ceramides Have Opposite Effects on Human Breast and Colon Cancer Cell Growth. Int. J. Biochem. Cell Biol. 2012, 44, 620–628.
- Schiffmann, S.; Hartmann, D.; Fuchs, S.; Birod, K.; Ferreirs, N.; Schreiber, Y.; Zivkovic, A.; Geisslinger, G.; Grösch, S.; Stark, H. Inhibitors of Specific Ceramide Synthases. Biochimie 2012, 94, 558–565.
- Verlekar, D.; Wei, S.J.; Cho, H.; Yang, S.; Kang, M.H. Ceramide Synthase-6 Confers Resistance to Chemotherapy by Binding to CD95/Fas in T-Cell Acute Lymphoblastic Leukemia. Cell Death Dis. 2018, 9, 1–14.
- Bose, R.; Verheij, M.; Haimovitz-friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide Synthase Mediates Daunorubicin-Lnduced Apoptosis: An Alternative Mechanism for Generating Death Signals. Cell 1995, 82, 405–414.
- Cabot, M.C.; Han, T.; Giuliano, A.E. The Multidrug Resistance Modulator SDZ PSC 833 Is a Potent Activator of Cellular Ceramide Formation. FEBS Lett. 1998, 431, 185–188.
- Charles, A.G.; Han, T.Y.; Liu, Y.Y.; Hansen, N.; Giuliano, A.E.; Cabot, M.C. Taxol-Induced Ceramide Generation and Apoptosis in Human Breast Cancer Cells. Cancer Chemother. Pharmacol. 2001, 47, 444–450.
- Fekry, B.; Esmaeilniakooshkghazi, A.; Krupenko, S.A.; Krupenko, N.I. Ceramide Synthase 6 Is a Novel Target of Methotrexate Mediating Its Antiproliferative Effect in a P53-Dependent Manner. PLoS ONE 2016, 11, e0146618.
- Lu, P.; White-Gilbertson, S.; Beeson, G.; Beeson, C.; Ogretmen, B.; Norris, J.; Voelkel-Johnson, C. Ceramide Synthase 6 Maximizes P53 Function to Prevent Progeny Formation from Polyploid Giant Cancer Cells. Cancers 2021, 13, 2212.
- Boppana, N.B.; Kodiha, M.; Stochaj, U.; Lin, H.S.; Haimovitz-Friedman, A.; Bielawska, A.; Bielawski, J.; Divine, G.W.; Boyd, J.A.; Korbelik, M.; et al. Ceramide Synthase Inhibitor Fumonisin B1 Inhibits Apoptotic Cell Death in SCC17B Human Head and Neck Squamous Carcinoma Cells after Pc4 Photosensitization. Photochem. Photobiol. Sci. 2014, 13, 1621–1627.
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of Ceramide Synthesis Ameliorates Glucocorticoid-, Saturated-Fat-, and Obesity-Induced Insulin Resistance. Cell Metab. 2007, 5, 167–179.
- Kraveka, J.M.; Li, L.; Szulc, Z.M.; Bielawski, J.; Ogretmen, B.; Hannun, Y.A.; Obeid, L.M.; Bielawska, A. Involvement of Dihydroceramide Desaturase in Cell Cycle Progression in Human Neuroblastoma Cells. J. Biol. Chem. 2007, 282, 16718–16728.
- Casasampere, M.; Ordóñez, Y.F.; Casas, J.; Fabrias, G. Dihydroceramide Desaturase Inhibitors Induce Autophagy via Dihydroceramide-Dependent and Independent Mechanisms. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 264–275.
- Hernández-Tiedra, S.; Fabriàs, G.; Dávila, D.; Salanueva, Í.J.; Casas, J.; Montes, L.R.; Antón, Z.; García-Taboada, E.; Salazar-Roa, M.; Lorente, M.; et al. Dihydroceramide Accumulation Mediates Cytotoxic Autophagy of Cancer Cells via Autolysosome Destabilization. Autophagy 2016, 12, 2213–2229.
- Missiroli, S.; Bonora, M.; Patergnani, S.; Poletti, F.; Perrone, M.; Gafà, R.; Magri, E.; Raimondi, A.; Lanza, G.; Tacchetti, C.; et al. PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development. Cell Rep. 2016, 16, 2415–2427.
- Signorelli, P.; Munoz-Olaya, J.M.; Gagliostro, V.; Casas, J.; Ghidoni, R.; Fabriàs, G. Dihydroceramide Intracellular Increase in Response to Resveratrol Treatment Mediates Autophagy in Gastric Cancer Cells. Cancer Lett. 2009, 282, 238–243.
- Ordóñez, Y.F.; González, J.; Bedia, C.; Casas, J.; Abad, J.L.; Delgado, A.; Fabrias, G. 3-Ketosphinganine Provokes the Accumulation of Dihydroshingolipids and Induces Autophagy in Cancer Cells. Mol. Biosyst. 2016, 12, 1166–1173.
- Boppana, N.B.; Delor, J.S.; van Buren, E.; Bielawska, A.; Bielawski, J.; Pierce, J.S.; Korbelik, M.; Separovic, D. Enhanced Apoptotic Cancer Cell Killing after Foscan Photodynamic Therapy Combined with Fenretinide via de Novo Sphingolipid Biosynthesis Pathway. J. Photochem. Photobiol. B Biol. 2016, 159, 191–195.
- Cataldi, S.; Borrelli, A.; Ceccarini, M.R.; Nakashidze, I.; Codini, M.; Belov, O.; Ivanov, A.; Krasavin, E.; Ferri, I.; Conte, C.; et al. Acid and Neutral Sphingomyelinase Behavior in Radiation-Induced Liver Pyroptosis and in the Protective/Preventive Role of RMnSOD. Int. J. Mol. Sci. 2020, 21, 3281.
- Malhi, H.; Guicciardi, M.E.; Gores, G.J. Hepatocyte Death: A Clear and Present Danger. Physiol. Rev. 2010, 90, 1165–1194.
- Clarke, C.J.; Cloessner, E.A.; Roddy, P.L.; Hannun, Y.A. Neutral Sphingomyelinase 2 (NSMase 2) Isthe Primary Neutral Sphingomyelinase Isoform Activated by Tumour Necrosis Factor-α in MCF-7 Cells. Biochem. J. 2011, 435, 381–390.
- Shamseddine, A.; Clarke, C.; Carroll, B.; Airola, M.V.; Mohammed, S.; Rella, A.; Obeid, L.; Hannun, Y. P53-Dependent Upregulation of Neutral Sphingomyelinase-2: Role in Doxorubicin-Induced Growth Arrest. Cell Death Dis. 2015, 6, e1947.
- Kachler, K.; Bailer, M.; Heim, L.; Schumacher, F.; Reichel, M.; Holzinger, C.D.; Trump, S.; Mittler, S.; Monti, J.; Trufa, D.I.; et al. Enhanced Acid Sphingomyelinase Activity Drives Immune Evasion and Tumor Growth in Non–Small Cell Lung Carcinoma. Cancer Res. 2017, 77, 5963–5976.
- Carpinteiro, A.; Becker, K.A.; Japtok, L.; Hessler, G.; Keitsch, S.; Požgajovà, M.; Schmid, K.W.; Adams, C.; Müller, S.; Kleuser, B.; et al. Regulation of Hematogenous Tumor Metastasis by Acid Sphingomyelinase. EMBO Mol. Med. 2015, 7, 714–734.
- Mizutani, N.; Omori, Y.; Kawamoto, Y.; Sobue, S.; Ichihara, M.; Suzuki, M.; Kyogashima, M.; Nakamura, M.; Tamiya-Koizumi, K.; Nozawa, Y.; et al. Resveratrol-Induced Transcriptional up-Regulation of ASMase (SMPD1) of Human Leukemia and Cancer Cells. Biochem. Biophys. Res. Commun. 2016, 470, 851–856.
- Kornhuber, J.; Tripal, P.; Reichel, M.; Mühle, C.; Rhein, C.; Muehlbacher, M.; Groemer, T.W.; Gulbins, E. Functional Inhibitors of Acid Sphingomyelinase (FIASMAS): A Novel Pharmacological Group of Drugs with Broad Clinical Applications. Cell. Physiol. Biochem. 2010, 26, 9–20.
- Carpinteiro, A.; Edwards, M.J.; Hoffmann, M.; Ahmad, S.A.; Fassbender, K.; Gulbins, E. Pharmacological Inhibition of Acid Sphingomyelinase Prevents Uptake of SARS-CoV-2 by Epithelial Cells. Cell Rep. Med. 2020, 1, 100142.
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the Endolysosomal Host-SARS-CoV-2 Interface by Clinically Licensed Functional Inhibitors of Acid Sphingomyelinase (FIASMA) Including the Antidepressant Fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255.
- le Corre, P.; Loas, G. Repurposing Functional Inhibitors of Acid Sphingomyelinase (Fiasmas): An Opportunity against SARS-CoV-2 Infection? J. Clin. Pharm. Ther. 2021, 1–7.
- Horinouchi, K.; Erlich, S.; Perl, D.P.; Ferlinz, K.; Bisgaier, C.L.; Sandhoff, K.; Desnick, R.J.; Stewart, C.L.; Schuchman, E.H. Acid Sphingomyelinase Deficient Mice: A Model of Types A and B Niemann–Pick Disease. Nat. Genet. 1995, 10, 288–293.
- Kuzu, O.F.; Gowda, R.; Noory, M.A.; Robertson, G.P. Modulating Cancer Cell Survival by Targeting Intracellular Cholesterol Transport. Br. J. Cancer 2017, 117, 513–524.
- Naser, E.; Kadow, S.; Schumacher, F.; Mohamed, Z.H.; Kappe, C.; Hessler, G.; Pollmeier, B.; Kleuser, B.; Arenz, C.; Becker, K.A.; et al. Characterization of the Small Molecule ARC39, a Direct and Specific Inhibitor of Acid Sphingomyelinase in Vitro. J. Lipid Res. 2020, 61, 896–910.
- Prause, K.; Naseri, G.; Schumacher, F.; Kappe, C.; Kleuser, B.; Arenz, C. A Photocaged Inhibitor of Acid Sphingomyelinase. Chem. Commun. 2020, 56, 14885–14888.
- Börtlein, C.; Schumacher, F.; Kleuser, B.; Dölken, L.; Avota, E. Role of Neutral Sphingomyelinase-2 (Nsm 2) in the Control of t Cell Plasma Membrane Lipid Composition and Cholesterol Homeostasis. Front. Cell Dev. Biol. 2019, 7, 226.
- Mizrachi, A.; Ben-Aharon, I.; Li, H.; Bar-Joseph, H.; Bodden, C.; Hikri, E.; Popovtzer, A.; Shalgi, R.; Haimovitz-Friedman, A. Chemotherapy-Induced Acute Vascular Injury Involves Intracellular Generation of ROS via Activation of the Acid Sphingomyelinase Pathway. Cell. Signal. 2021, 82, 109969.
- Rajagopal, C.; Harikumar, K.B. The Origin and Functions of Exosomes in Cancer. Front. Oncol. 2018, 8, 66.
- Kunou, S.; Shimada, K.; Takai, M.; Sakamoto, A.; Aoki, T.; Hikita, T.; Kagaya, Y.; Iwamoto, E.; Sanada, M.; Shimada, S.; et al. Exosomes Secreted from Cancer-Associated Fibroblasts Elicit Anti-Pyrimidine Drug Resistance through Modulation of Its Transporter in Malignant Lymphoma. Oncogene 2021, 40, 3989–4003.
- Li, S.; Yi, M.; Dong, B.; Jiao, Y.; Luo, S.; Wu, K. The Roles of Exosomes in Cancer Drug Resistance and Its Therapeutic Application. Clin. Transl. Med. 2020, 10, e257.
- Kosaka, N.; Iguchi, H.; Hagiwara, K.; Yoshioka, Y.; Takeshita, F.; Ochiya, T. Neutral Sphingomyelinase 2 (NSMase2)-Dependent Exosomal Transfer of Angiogenic Micrornas Regulate Cancer Cell Metastasis. J. Biol. Chem. 2013, 288, 10849–10859.
- Lin, M.; Liao, W.; Dong, M.; Zhu, R.; Xiao, J.; Sun, T.; Chen, Z.; Wu, B.; Jin, J. Exosomal Neutral Sphingomyelinase 1 Suppresses Hepatocellular Carcinoma via Decreasing the Ratio of Sphingomyelin/Ceramide. FEBS J. 2018, 285, 3835–3848.
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Kim, J.H. Platinum Nanoparticles Enhance Exosome Release in Human Lung Epithelial Adenocarcinoma Cancer Cells (A549): Oxidative Stress and the Ceramide Pathway Are Key Players. Int. J. Nanomed. 2021, 16, 515–538.
- Chen, Y.; Cao, Y. The Sphingomyelin Synthase Family: Proteins, Diseases, and Inhibitors. Biol. Chem. 2017, 398, 1319–1325.
- Gulbins, A.; Schumacher, F.; Becker, K.A.; Wilker, B.; Soddemann, M.; Boldrin, F.; Müller, C.P.; Edwards, M.J.; Goodman, M.; Caldwell, C.C.; et al. Antidepressants Act by Inducing Autophagy Controlled by Sphingomyelin–Ceramide. Mol. Psychiatry 2018, 23, 2324–2346.
- Barceló-Coblijn, G.; Martin, M.L.; de Almeida, R.F.M.; Noguera-Salvà, M.A.; Marcilla-Etxenike, A.; Guardiola-Serrano, F.; Lüth, A.; Kleuser, B.; Halver, J.E.; Escribá, P.v. Sphingomyelin and Sphingomyelin Synthase (SMS) in the Malignant Transformation of Glioma Cells and in 2-Hydroxyoleic Acid Therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19569–19574.
- Deng, Y.; Hu, J.C.; He, S.H.; Lou, B.; Ding, T.B.; Yang, J.T.; Mo, M.G.; Ye, D.Y.; Zhou, L.; Jiang, X.C.; et al. Sphingomyelin Synthase 2 Facilitates M2-like Macrophage Polarization and Tumor Progression in a Mouse Model of Triple-Negative Breast Cancer. Acta Pharmacol. Sin. 2021, 42, 149–159.
- Jing, F.; Jing, C.; Dai, X.; Zhou, G.; Di, S.; Bi, X.; Dai, T.; Qin, T.; Hong, L. Sphingomyelin Synthase 2 but Not Sphingomyelin Synthase 1 Is Upregulated in Ovarian Cancer and Involved in Migration, Growth and Survival via Different Mechanisms. Am. J. Transl. Res. 2021, 13, 4412–4421.
- Zheng, K.; Chen, Z.; Feng, H.; Chen, Y.; Zhang, C.; Yu, J.; Luo, Y.; Zhao, L.; Jiang, X.; Shi, F. Sphingomyelin Synthase 2 Promotes an Aggressive Breast Cancer Phenotype by Disrupting the Homoeostasis of Ceramide and Sphingomyelin. Cell Death Dis. 2019, 10, 1–11.
- Bilal, F.; Montfort, A.; Gilhodes, J.; Garcia, V.; Riond, J.; Carpentier, S.; Filleron, T.; Colacios, C.; Levade, T.; Daher, A.; et al. Sphingomyelin Synthase 1 (SMS1) Downregulation Is Associated with Sphingolipid Reprogramming and a Worse Prognosis in Melanoma. Front. Pharmacol. 2019, 10, 443.
- Song, M.; Zang, W.; Zhang, B.; Cao, J.; Yang, G. GCS Overexpression Is Associated with Multidrug Resistance of Human HCT-8 Colon Cancer Cells. J. Exp. Clin. Cancer Res. 2012, 31, 1–6.
- Liu, J.; Wang, S.; Wang, C.; Kong, X.; Sun, P. Prognostic Value of Using Glucosylceramide Synthase and Cytochrome P450 Family 1 Subfamily A1 Expression Levels for Patients with Triple-negative Breast Cancer Following Neoadjuvant Chemotherapy. Exp. Ther. Med. 2021, 21, 1.
- Lee, J.Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.J.; et al. Polyunsaturated Fatty Acid Biosynthesis Pathway Determines Ferroptosis Sensitivity in Gastric Cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 32433–32442.
- Gouazé, V.; Liu, Y.Y.; Prickett, C.S.; Yu, J.Y.; Giuliano, A.E.; Cabot, M.C. Glucosylceramide Synthase Blockade Down-Regulates P-Glycoprotein and Resensitizes Multidrug-Resistant Breast Cancer Cells to Anticancer Drugs. Cancer Res. 2005, 65, 3861–3867.
- Madigan, J.P.; Robey, R.W.; Poprawski, J.E.; Huang, H.; Clarke, C.J.; Gottesman, M.M.; Cabot, M.C.; Rosenberg, D.W. A Role for Ceramide Glycosylation in Resistance to Oxaliplatin in Colorectal Cancer. Exp. Cell Res. 2020, 388, 111860.
- Snider, A.J.; Seeds, M.C.; Johnstone, L.; Snider, J.M.; Hallmark, B.; Dutta, R.; Franco, C.M.; Parks, J.S.; Bensen, J.T.; Broeckling, C.D.; et al. Identification of Plasma Glycosphingolipids as Potential Biomarkers for Prostate Cancer (PCA) Status. Biomolecules 2020, 10, 1393.
- Hartwig, P.; Höglinger, D. The Glucosylceramide Synthase Inhibitor PDMP Causes Lysosomal Lipid Accumulation and MTOR Inactivation. Int. J. Mol. Sci. 2021, 22, 7065.
- Ortega, R.A.; Wang, C.; Raymond, D.; Bryant, N.; Scherzer, C.R.; Thaler, A.; Alcalay, R.N.; West, A.B.; Mirelman, A.; Kuras, Y.; et al. Association of Dual LRRK2 G2019S and GBA Variations with Parkinson Disease Progression. JAMA Netw. Open 2021, 4, 215845.
- Gan-Or, Z.; Liong, C.; Alcalay, R.N. GBA-Associated Parkinson’s Disease and Other Synucleinopathies. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–10.
- Astudillo, L.; Therville, N.; Colacios, C.; Ségui, B.; Andrieu-Abadie, N.; Levade, T. Glucosylceramidases and Malignancies in Mammals. Biochimie 2016, 125, 267–280.
- Sturchio, A.; Dwivedi, A.K.; Vizcarra, J.A.; Chirra, M.; Keeling, E.G.; Mata, I.F.; Kauffman, M.A.; Pandey, M.K.; Roviello, G.; Comi, C.; et al. Genetic Parkinsonisms and Cancer: A Systematic Review and Meta-Analysis. Rev. Neurosci. 2021, 32, 159–167.
- Li, Z.; Xu, D.; Tong, X.; Shan, C. Inhibition of β-Glucosidase Overcomes Gastric Cancer Chemoresistance through Inducing Lysosomal Dysfunction. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101456.
- Coant, N.; Hannun, Y.A. Neutral Ceramidase: Advances in Mechanisms, Cell Regulation, and Roles in Cancer. Adv. Biol. Regul. 2019, 71, 141–146.
- Xu, R.; Antwi Boasiako, P.; Mao, C. Alkaline Ceramidase Family: The First Two Decades. Cell. Signal. 2021, 78, 109860.
- Vijayan, Y.; Lankadasari, M.B.; Harikumar, K.B. Acid Ceramidase: A Novel Therapeutic Target in Cancer. Curr. Top. Med. Chem. 2019, 19, 1512–1520.
- Saied, E.M.; Arenz, C. Inhibitors of Ceramidases. Chem. Phys. Lipids 2016, 197, 60–68.
- Lai, M.; La Rocca, V.; Amato, R.; Freer, G.; Costa, M.; Spezia, P.G.; Quaranta, P.; Lombardo, G.; Piomelli, D.; Pistello, M. Ablation of Acid Ceramidase Impairs Autophagy and Mitochondria Activity in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 3247.
- Lai, M.; Amato, R.; La Rocca, V.; Bilgin, M.; Freer, G.; Spezia, P.; Quaranta, P.; Piomelli, D.; Pistello, M. Acid Ceramidase Controls Apoptosis and Increases Autophagy in Human Melanoma Cells Treated with Doxorubicin. Sci. Rep. 2021, 11, 1–14.
- Clifford, R.E.; Govindarajah, N.; Bowden, D.; Sutton, P.; Glenn, M.; Darvish-Damavandi, M.; Buczacki, S.; McDermott, U.; Szulc, Z.; Ogretmen, B.; et al. Targeting Acid Ceramidase to Improve the Radiosensitivity of Rectal Cancer. Cells 2020, 9, 2693.
- White-Gilbertson, S.; Lu, P.; Jones, C.M.; Chiodini, S.; Hurley, D.; Das, A.; Delaney, J.R.; Norris, J.S.; Voelkel-Johnson, C. Tamoxifen Is a Candidate First-in-Class Inhibitor of Acid Ceramidase That Reduces Amitotic Division in Polyploid Giant Cancer Cells—Unrecognized Players in Tumorigenesis. Cancer Med. 2020, 9, 3142–3152.
- Rothemund, M.; Bär, A.; Klatt, F.; Weidler, S.; Köhler, L.; Unverzagt, C.; Kuhn, C.D.; Schobert, R. N-Metallocenoylsphingosines as Targeted Ceramidase Inhibitors: Syntheses and Antitumoral Effects. Bioorg. Chem. 2020, 97, 103703.
- Liu, B.; Xiao, J.; Dong, M.; Qiu, Z.; Jin, J. Human Alkaline Ceramidase 2 Promotes the Growth, Invasion, and Migration of Hepatocellular Carcinoma Cells via Sphingomyelin Phosphodiesterase Acid-like 3B. Cancer Sci. 2020, 111, 2259.
- Zhang, S.; Huang, P.; Dai, H.; Li, Q.; Hu, L.; Peng, J.; Jiang, S.; Xu, Y.; Wu, Z.; Nie, H.; et al. TIMELESS Regulates Sphingolipid Metabolism and Tumor Cell Growth through Sp1/ACER2/S1P Axis in ER-Positive Breast Cancer. Cell Death Dis. 2020, 11, 1–14.
- Yin, Y.; Xu, M.; Gao, J.; Li, M. Alkaline Ceramidase 3 Promotes Growth of Hepatocellular Carcinoma Cells via Regulating S1P/S1PR2/PI3K/AKT Signaling. Pathol. Res. Pract. 2018, 214, 1381–1387.
- Steiner, R.; Saied, E.M.; Othman, A.; Arenz, C.; Maccarone, A.T.; Poad, B.L.J.; Blanksby, S.J.; von Eckardstein, A.; Hornemann, T. Elucidating the Chemical Structure of Native 1-Deoxysphingosine. J. Lipid Res. 2016, 57, 1194–1203.
- Saied, E.M.; Le, T.L.S.; Hornemann, T.; Arenz, C. Synthesis and Characterization of Some Atypical Sphingoid Bases. Bioorg. Med. Chem. 2018, 26, 4047–4057.
- Bielsa, N.; Casasampere, M.; Aseeri, M.; Casas, J.; Delgado, A.; Abad, J.L.; Fabriàs, G. Discovery of Deoxyceramide Analogs as Highly Selective ACER3 Inhibitors in Live Cells. Eur. J. Med. Chem. 2021, 216, 113296.
- Vasiliauskaité-Brooks, I.; Healey, R.D.; Rochaix, P.; Saint-Paul, J.; Sounier, R.; Grison, C.; Waltrich-Augusto, T.; Fortier, M.; Hoh, F.; Saied, E.M.; et al. Structure of a Human Intramembrane Ceramidase Explains Enzymatic Dysfunction Found in Leukodystrophy. Nat. Commun. 2018, 9, 1–13.