Circadian clocks control several homeostatic processes in mammals through internal molecular mechanisms. Chronic perturbation of circadian rhythms is associated with metabolic diseases and increased cancer risk, including liver cancer. The hepatic physiology follows a daily rhythm, driven by clock genes that control the expression of several proteins involved in distinct metabolic pathways. Alteration of the liver clock results in metabolic disorders, such as non-alcoholic fatty liver diseases (NAFLD) and impaired glucose metabolism, that can trigger the activation of oncogenic pathways, inducing spontaneous hepatocarcinoma (HCC). The study of oncogenic changes related to alterations of liver clock provides new potentially useful targets for prevention and management of HCC.
- circadian clock
- liver
- hepatocellular carcinoma
1. Introduction to Circadian Rhythms
Circadian rhythms in mammals allow tissues to adapt to their biological function and to anticipate external changes, which leads to the synchronization of mammal physiology to the environment and to the 24-h solar day. These daily fluctuations in physiological and behavioral processes rely on an intrinsic molecular clock and its interaction with environmental changes. The external cues that dictate circadian behaviors are known as zeitgebers. The light/dark cycle is the most studied and common zeitgeber, but many other signals, including the feeding/fasting cycle, melatonin or body temperature, are considered zeitgebers as well [1]. Disruption of circadian rhythms in mammals can result in the development of different diseases, such as cancer and neurodegenerative and metabolic disorders. Therefore, it is fundamental to study the molecular mechanisms that sustain these circadian rhythms in order to better understand how disease can originate from abruptions in daily fluctuations.
The circadian system in mammals relies on the presence of a molecular clock in every organ and every cell of the body, thus generating multiple autonomous clocks. All of these self-sustaining clocks are synchronized by a central molecular clock within the suprachiasmatic nucleus (SCN), composing a hierarchically structured circadian system. This master synchronizer is located in the anteroventral hypothalamus, where it receives external photic information derived from the retina as well as non-photic information from the other tissues and organs of the body. The SCN integrates all of this information and generates output signals to the peripheral clocks, setting the basis for an entrainment between the tissues as well as to the 24-h day. There are three major sources of input signals arriving at the SCN: (i) the photic information comes from the transduction of the light by photosensitive retinal cells, which transmit this photic information to neurons in the SCN by releasing melanopsin; (ii) melanopsin then acts on another region of the brain, the intergeniculate leaflets (IGL), to instruct the release of neurotransmitters from these neurons to the SCN. These two mechanisms support both a direct and an indirect entrainment of SCN to light, with a delay between both inputs that makes the SCN response more precise and determinant; and (iii) non-photic information, such as that related to activity and exercise, arrive at the SCN from the median/dorsal raphe nucleus (MRN/DRN) [1].
Although peripheral clocks act with certain autonomy, their orchestration via the SCN is necessary to coordinate a network between peripheral clocks [2]. SCN coordinates peripheral clocks directly through humoral and synaptic signals. For instance, sympathetic denervation of some peripheral clocks results in altered circadian oscillations [3], and glucocorticoids are released in a circadian manner to phase-entrain peripheral oscillators [4]. Further, by coupling feeding–fasting and rest–activity cycles, the SCN can indirectly dictate daily fluctuations in peripheral tissues. Normally, direct and indirect pathways cooperate to entrain the peripheral timekeepers. However, time-restriction feeding is able to desynchronize peripheral clocks from the SCN without affecting the rhythmicity of the central pacemaker, indicating that the feeding–fasting cycle is a dominant zeitgeber in peripheral clocks [5]. Interestingly, body temperature also modulates self-sustaining clocks independently of the SCN [6]. The existence of these extra-SCN pacemakers that are able to coordinate clocks in peripheral tissues raised the intriguing questions of how independent these autonomous clocks actually are from the SCN, and whether this network can be maintained efficiently without central pacemaker contribution [7].
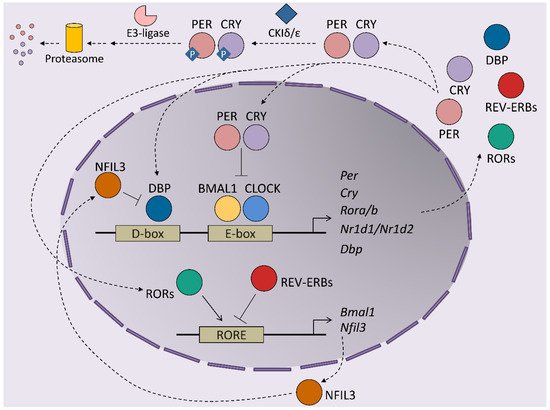
At the molecular level, circadian rhythms are orchestrated by transcription/translation feedback loops (TTFL) of activator and repressor components of the molecular clock ( Figure 1 ). The activator components are two bHLH-PAS proteins, BMAL1 and CLOCK, which form heterodimers that bind E-box regions on the promoters of their target genes [8]. This set of genes includes those that encode the repressor components of the clock, cryptochrome ( Cry1 and Cry2 ) and the Period proteins ( Per1 , Per2 and Per3 ). The protein products of these genes dimerize and form complexes between themselves, which allows them to translocate into the nucleus, where they inhibit the transactivation activity of the BMAL1-CLOCK complex, thereby repressing their own expression [9][10][11]. BMAL1-CLOCK activity begins at the start of the circadian day, driving the accumulation of CRY and PER transcripts and proteins during the second part of the day. PER and CRY repressor complexes inhibit BMAL1-CLOCK activity in the nucleus during the night by repressing their own expression. As repression progresses, post-translational modifications of CRY and PER allow their inhibition and degradation by the proteasome. Most of these modifications rely on inhibitory phosphorylation by CKIε/δ and on ubiquitination by E3 ligases [12][13][14]. Turnover of the CRY and PER proteins restores BMAL1-CLOCK complexes to an active state, making them available to restart the cycle. This autoregulatory feedback loop is also maintained by the orphan nuclear receptors, REV-ERBs and RORs. Both receptors are activated by BMAL1-CLOCK and compete for binding to RORs in the promoter of their target genes, where they exert opposing roles. Namely, REV-ERBs repress BMAL1 expression, while RORs induces Bmal1 transcription, contributing to the restarting of the clock [15][16]. This REV-ERB –ROR loop rhythm is further maintained by the DBP and NFIL3 transcription factors, which also exert activator and repressor functions, respectively, in ROR-encoding genes [17]. Post-translational regulation of the clock components is essential for maintaining the delay between the positive and negative limbs of the clock. In addition to Cry and Per post-translational regulation, phosphorylation is crucial for BMAL1 activity: phosphorylation by CKIε positively regulates BMAL1 and enhances the transcriptional activity of the BMAL1-CLOCK complex, while MAPKs phosphorylate and thereby negatively regulate BMAL1 protein [18][19]. In the particular case of the liver, around 25% of all proteins show circadian oscillation in their phosphorylation status, highlighting this post-transcriptional modification as a main timekeeper in hepatic cells [20]. In fact, this molecular oscillator not only regulates the expression of the core clock genes but also the transcription of many protein-coding genes in all tissues, thereby driving their rhythmic expression during the 24-h day cycle. Indeed, between 5 and 20% of all mouse transcripts show circadian fluctuations in any given tissue, whereby the liver is the organ that has the most fluctuations [21]. These results highlight the role of clock genes in the transmission of circadian information to non-clock proteins, integrating cellular physiology and circadian fluctuations. Indeed, the circadian output of a cell results from the interflow between the intrinsic TTFL molecular regulation of clock genes, cellular metabolism and external cues. This plasticity of the molecular clock allows cellular circadian transcriptome to adapt to these external changes [22].
2. Modulators of Hepatic Circadian Rhythms
Liver homeostasis requires the tight circadian regulation of its metabolism, and glucose, bile acids, lipids and cholesterol are all subject to a timed circadian control [23]. This daily control of liver metabolism must adapt to changes in nutrition regimes [24][25]. The regulation of liver metabolism by feeding was demonstrated using the luciferase system in vivo. Specifically, it was shown that restricted feeding could rapidly entrain the liver circadian clock [23]. The relevance of feeding cycles for the liver clock was demonstrated by showing that feeding regimes could improve circadian gene expression patterns in mice deficient in CRY proteins [26].
The type of diet also affects the circadian liver clock; for instance, a high-fat diet (HFD) generates a profound reorganization of hepatic metabolic pathways and remodeling of the liver clock [23]. Interestingly, this reorganization of liver circadian metabolism is independent of obesity and is mediated by direct effects of the diet [25]. Recently, our group demonstrated that part of this reprogramming of liver circadian rhythm is caused by the deregulation of neutrophils infiltration in the liver, for instance from diet or jet lag [27]. Neutrophils show a circadian infiltration into the liver, where they signal hepatocytes through the secretion of the serine protease elastase, synchronizing the circadian clock and liver metabolism. However, both jet lag and unhealthy diet (for instance, deficient in methionine and choline, which leads to NAFLD, or high-fat) have been shown to disrupt the circadian neutrophil infiltration into the liver, resulting in alterations of the daily hepatic metabolism. Additionally, a ketogenic diet (KD), which is a popular high-fat, low-carbohydrate diet regime used to lose weight [28], also has profound effects on the liver clock, and especially on BMAL1-controlled genes [29].
The regulation of liver circadian rhythm is, in part, mediated by the liver-specific clock machinery, as ablation of circadian transcription genes results in dysregulation of the liver metabolic clock, including loss of oscillation in the glucose, lipid and oxidative pathways [30][31]. The intrinsic control of the liver circadian clock was demonstrated using conditional animals in which clock genes were removed only in hepatocytes. Mice lacking hepatic BMAL1 presented hypoglycemia during the day, due to an exacerbated glucose clearance, and disruption of circadian expression of glucose regulatory genes in the liver [30]. In addition, an intrinsic control of the liver clock—even in the absence of external signal or other circadian clocks—has been demonstrated using animals in which only hepatocytes express BMAL1 [32][33]. Although the key role of BMAL1 in the regulation of circadian liver metabolism is clear, it has recently been demonstrated that Bmal1 −/− mice maintain circadian oscillations in their transcriptome, proteome and phosphoproteome even in the absence of any exogenous drivers, such as daily light or temperature cycles [34]. These data suggest that there are other players that keep this daily regulation. Besides this transcriptional regulation of metabolism, circulating hormones and the activation of several signaling pathways also participate in the circadian regulation [31]. The hormone-responsive nuclear receptors REV-ERBs and RORs are important liver clock components that regulate metabolism [35]. In mice, deletion of hepatic Rev-erbs decreases the diurnal rhythm of the clock genes Bmal1 and Clock and enhances the rhythmic amplitudes of genes implicated in lipid metabolism, thereby affecting the diurnal rhythm of de novo lipogenesis [36]. Synchronization between clock genes oscillation and the expression of different enzymes and metabolic master regulators is needed to organize daily hepatic metabolism [37]. Although the circadian phases of messenger RNAs are in concordance with their output protein expression, proteomic studies have found that despite the cycling of some metabolic enzymes, their transcripts are relatively constant throughout the day [38]. In addition to circadian protein expression, circadian phosphorylation has been demonstrated to control the daily cycle of liver metabolism [20]. Moreover, acetylation and methylation are important modulators of the liver circadian rhythm. In fact, CLOCK is a histone acetyltransferase that catalyzes the acetylation of BMAL1 [39].
In rodents, cholesterol and bile acid synthesis is synchronized with the timing of food intake [40]. While bile acid metabolism presents daily fluctuations in mice, in humans, cholesterol and bile acid synthesis are out-of-phase. For instance, in rodents, the rate-limiting enzyme for the synthesis of bile acids, cholesterol 7α-hydroxylase (CYP7A1), shows an early peak in the active phase [41][42]. Bile acid metabolism is, in part, regulated by several nuclear receptors, including the farnesoid-X receptor (FXR) and the liver receptor homolog 1 (LRH). FXR induces the negative co-repressor small heterodimer partner (SHP) to suppress CYP7A1 activity [23]. The clock gene Rev - erbα activates Cyp7a1 gene expression, possibly through the inhibition of Shp transcription [43]. We recently demonstrated that, in mice, the liver c-Jun NH 2-terminal kinases (JNK)1/2 have a circadian regulation in the liver [27] and control bile acid production; correspondingly, ablation of hepatic JNK1/2 results in bile acid dysregulation and cholestasis [44]. This alteration in the control of bile acid metabolism also appears in mice lacking the clock gene Per1/2, which present high levels of serum and liver bile acids and liver damage [45]. Overall, the crucial role of bile acids in the regulation of cholesterol metabolism prompts reconsideration of the possible contribution of bile acid circadian disruption to the hyperlipidemia found in shift workers [46].
3. Circadian Rhythms and Hepatic Metabolism
The liver is a central metabolic organ that governs whole-body homeostasis, and circadian rhythms play a major role in liver homeostasis, including hepatic metabolism. In fact, over 50% of liver metabolites have a circadian rhythm that is coupled with clock genes transcription [47]. Although the liver is influenced by central and other peripheral clocks, around 10% of normally rhythmic transcripts are expressed in an autonomous manner [32]. In this circadian molecular machinery, the hepatic clock–metabolism crosstalk seems to be regulated by a circadian phosphorylation of a variety of signaling pathways involved in several metabolic processes, including the MAPK and mTOR pathways [20]. Additionally, genetic or environmental disruption of the circadian clock triggers metabolic diseases or aggravates liver pathologies, supporting the main role of clock system in hepatic metabolism.
On the other hand, the role of BMAL1 in PPARα is not restricted to the AKT-mTOR pathway. It has been shown that liver-specific deletion of Bmal1 in mice leads to the accumulation of ROS in the liver, which increases mRNA methylation and results in the downregulation of the transcription of PPARα, thereby impacting the hepatic liver metabolism [48]. The specific ablation of Rev-erbα and Rev-erbβ in hepatocytes leads to alteration of the circadian expression of liver genes and results in the disruption of the daily rhythm of de novo lipogenesis [36]. Interestingly, treatment of obese mice with REV-ERB agonists modifies clock and metabolic gene expression, leading to reduced triglycerides and cholesterol synthesis in the liver [49]. These studies suggest that targeting the circadian clock may be a potential treatment for metabolic disorders.
Extrinsic factors, such as non-parenchymal cells, may also affect the liver circadian clock. We have recently published that neutrophils are able to control the liver circadian clock through the activation of hepatic JNKs [27]. JNKs activate Bmal1 transcription and repress the cycling hepatokine FGF21; both events affect liver lipid metabolism, by activating lipogenesis and reducing the FGF21-metabolic regulatory effect, respectively [27]. Interestingly, JNKs have a circadian activation in mouse liver [20], and disruption of hepatic JNKs disrupts bile acid production and results in cholangiocarcinoma [44]. This circadian regulation of kinase activation might mediate crosstalk between the circadian clock and metabolism. Moreover, hepatocytes circadian rhythm is able to reprogram daily changes in liver non-parenchymal cells. In fact, hepatic REV-ERBα/β modulates rhythmic transcriptomes and metabolomes of Kupffer, stellate and endothelial cells from the liver [36]. These findings are highly relevant as the liver function has been traditionally described as being influenced by non-parenchymal cells, and this is the first report outlining that it also operates in reverse, controlling the circadian rhythms.
Circadian liver metabolism is also controlled by a variety of proteins and key enzymes. For instance, some hepatokines (e.g., FGF21) and adipokines (e.g., adiponectin) have rhythmic expression patterns that affect the rhythmic patterns of liver metabolism. FGF21 plays important roles in adaptation to fasting, such as lipolysis and ketogenesis, and is rhythmically expressed in the presence of the BMAL1-CLOCK complex and RORα, and strongly repressed by E4BP4 [50]. Further, adiponectin, which is expressed by the adipose tissue with a circadian rhythm, plays a fundamental role in the regulation of insulin sensitivity [51]. Indeed, daily rhythmic variations of circulating adiponectin modulate liver glucose metabolism in humans [51].
4. Role of Circadian Rhythms in Liver Cancer
NAFLD and obesity are associated with people presenting alterations in sleep timing, such as night-shift workers or people with sleep dyspnea. This problem, commonly known as “social jet lag”, represents a risk factor for different metabolic disorders including cancer [52][53]. Several mouse models of circadian disruption and circadian genes knockout mouse models have allowed us to decipher the importance of circadian rhythmicity in HCC development in recent years ( Figure 2 ). Mechanistically, circadian disruption activates the constitutive aldosterone receptor (CAR) via sympathetic nervous system dysfunction and cholestasis, and this overexpressed CAR can promote liver tumor, which drives the progression from NAFLD to NASH and, ultimately, to HCC [54]. The pro-tumorigenic effect of circadian dysfunction had been also observed in previous studies. For instance, experimentally induced chronic jet lag in mice was shown to increase the frequency of tumor formation after exposure to the carcinogenic diethylnitrosamine (DEN), in part due to an increased expression of the c-Myc oncogene and a decreased expression of the tumor suppressor p53 [55]. In addition to the potentiation of the hepatic carcinogenic effect induced by DEN, chronic jet lag also provokes a variety of hepatic tumor types in mice, such as cholangiocarcinomas, sarcomas, mixed tumors and larger tumors [56]. Remarkably, DEN itself has the ability to disrupt circadian rhythms of rest–activity and body temperature in mice [57]. DEN circadian disruption suggests that part of its carcinogen effects are mediated by alterations in the hepatic daily cycle, and that circadian reprogramming might be critical for stopping hepatic cancer development. Indeed, chronic circadian disruption also acts as a pro-tumorigenic factor in livers from steroid receptor coactivator-2 (SRC-2) knockout mice. SRC-2 works as crucial integrator of circadian behavior governed by light and metabolic homeostasis sustained by the liver; notably, a lack of SRC-2 causes mice to be unable to adapt to the stress of chronic circadian disruption and provokes NASH and HCC gene signatures [58], demonstrating the fundamental role of circadian cycles in the development of HCC.
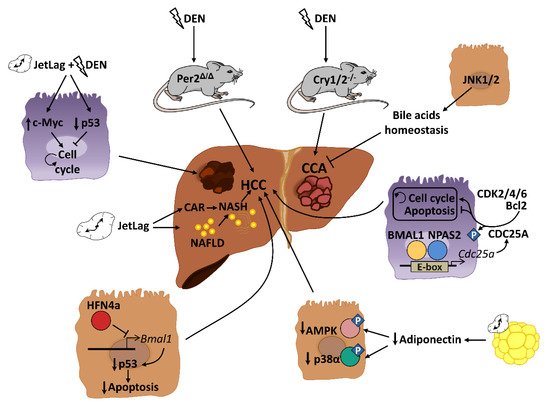
Genetic and epigenetic alterations of clock genes can also drive carcinogenesis in different mutant and knockout mouse models. PER2, CRY1 and CRY2 seem to be critical for controlling hepatic carcinogenesis [55][57]. A lack of PER2 increases cMyc expression and disrupts clock-controlled pathways and patterns [55][57]; consequently, these mice are more susceptible to developing HCC after DEN injection [57]. Full deletion of Cry1 and Cry2 also disrupts the molecular circadian clock and increases chemically induced liver carcinogenesis. However, in this case, CRY seems to affect the molecular pathways involved in bile duct carcinogenesis, as its deletion dramatically increases the frequency of cholangiocarcinoma after DEN exposure [59]. These studies are in agreement with published data in humans, in which comparing HCC samples to their corresponding adjacent normal tissue shows significant alterations in the circadian genes Per and Cry [60][61][62]. As mentioned above, mice with loss-of-function of the Clock gene spontaneously develop NAFLD in certain chow diet conditions (such as HFD), which can progress to steatohepatitis and cirrhosis with age [63]. The additional deletion of the apolipoprotein ApoE, whose deficiency increases the susceptibility to NALFD under HFD conditions, increases the liver pathology in these mice, including the augmented expression of cancer markers in the liver [63]. Analysis of miRNA profiles in liver from Clock mutant mice revealed that Clock-regulated miRNAs may be involved in cancer initiation or progression by controlling genes related to cell proliferation, invasion and/or metabolism in the mouse liver [64]. Thus, deregulation of the Clock function might predispose an organism to liver cancer development. Additionally, the neuronal PAS domain protein 2 (NPAS2), a core circadian molecule analog of CLOCK, is upregulated in HCC and facilitates cancer cell survival [65]. Mechanistically, NPAS2 heterodimerizes with BMAL1 and induces the expression of the phosphatase CDC25A, which dephosphorylates the cyclin-dependent kinases (CDK)2, CDK4 and CDK6 as well as Bcl-2, thereby promoting cell proliferation and inhibiting mitochondria-dependent intrinsic apoptosis, respectively [65]. Therefore, NPAS2 is an important contributor to poor prognosis of HCC and may constitute a potential therapeutic target in HCC patients. Further, although there are no available data about the role of BMAL1 in the HCC development, it has been demonstrated that one isoform of the tumor suppressor HNF4a, which was induced in some forms of HCC, can repress BMAL1 expression, and that forced expression of BMAL1 in HFN4a-positive HCC prevents the growth of tumors in vivo by stimulating p53 expression and the activation of apoptosis [66].
Disruption of circadian-controlled genes that have hepato-protective functions could be a potential cause of HCC. Circulating adiponectin levels, which show circadian oscillation [51], significantly prevent HCC development through the hepatic activation of p38α and the AMP-activated protein kinase (AMPK) [67]. Decreased levels of circulating adiponectin by circadian disruption could negatively affect this antitumor activity. Similar effects could be also observed in the rhythmic disturbance of FGF-21, which is essential for preventing NAFLD progression to HCC during long-term obesogenic diet [68].
All these data indicate that a correct function of the biological clock has tumor-suppressing potential, while disturbing the normal circadian rhythm is an important risk factor for HCC. Thus, manipulation of circadian rhythms might represent an important strategy to prevent the development of HCC and to design new strategies for HCC treatment.
This entry is adapted from the peer-reviewed paper 10.3390/cancers13143631
References
- Dibner, C.; Schibler, U.; Albrecht, U. The Mammalian Circadian Timing System: Organization and Coordination of Central and Peripheral Clocks. Annu. Rev. Physiol. 2010, 72, 517–549.
- Guo, H.; Brewer, J.M.; Lehman, M.N.; Bittman, E.L. Suprachiasmatic Regulation of Circadian Rhythms of Gene Expression in Hamster Peripheral Organs: Effects of Transplanting the Pacemaker. J. Neurosci. 2006, 26, 6406–6412.
- Vujović, N.; Davidson, A.J.; Menaker, M. Sympathetic input modulates, but does not determine, phase of peripheral circadian oscillators. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R355–R360.
- Balsalobre, A.; Brown, S.A.; Marcacci, L.; Tronche, F.; Kellendonk, C.; Reichardt, H.M.; Schütz, G.; Schibler, U. Resetting of Circadian Time in Peripheral Tissues by Glucocorticoid Signaling. Science 2000, 289, 2344–2347.
- Damiola, F.; Le Minh, N.; Preitner, N.; Kornmann, B.; Fleury-Olela, F.; Schibler, U. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 2000, 14, 2950–2961.
- Brown, S.A.; Zumbrunn, G.; Fleury-Olela, F.; Preitner, N.; Schibler, U. Rhythms of Mammalian Body Temperature Can Sustain Peripheral Circadian Clocks. Curr. Biol. 2002, 12, 1574–1583.
- Pezuk, P.; Mohawk, J.A.; Yoshikawa, T.; Sellix, M.T.; Menaker, M. Circadian Organization Is Governed by Extra-SCN Pacemakers. J. Biol. Rhythm. 2010, 25, 432–441.
- Gekakis, N.; Staknis, D.; Nguyen, H.B.; Davis, F.C.; Wilsbacher, L.D.; King, D.P.; Takahashi, J.S.; Weitz, C.J. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998, 280, 1564–1569.
- Sangoram, A.M.; Saez, L.; Antoch, M.P.; Gekakis, N.; Staknis, D.; Whiteley, A.; Fruechte, E.M.; Vitaterna, M.H.; Shimomura, K.; King, D.P.; et al. Mammalian Circadian Autoregulatory Loop: A Timeless Ortholog and mPer1 Interact and Negatively Regulate CLOCK-BMAL1-Induced Transcription. Neuron 1998, 21, 1101–1113.
- Kume, K.; Zylka, M.J.; Sriram, S.; Shearman, L.P.; Weaver, D.; Jin, X.; Maywood, E.S.; Hastings, M.H.; Reppert, S.M. mCRY1 and mCRY2 Are Essential Components of the Negative Limb of the Circadian Clock Feedback Loop. Cell 1999, 98, 193–205.
- Shearman, L.P.; Sriram, S.; Weaver, D.; Maywood, E.S.; Chaves, I.; Zheng, B.; Kume, K.; Lee, C.C.; Van Der Horst, G.T.; Hastings, M.H.; et al. Interacting Molecular Loops in the Mammalian Circadian Clock. Science 2000, 288, 1013–1019.
- Eide, E.J.; Woolf, M.F.; Kang, H.; Woolf, P.; Hurst, W.; Camacho, F.; Vielhaber, E.L.; Giovanni, A.; Virshup, D.M. Control of Mammalian Circadian Rhythm by CKIε-Regulated Proteasome-Mediated PER2 Degradation. Mol. Cell. Biol. 2005, 25, 2795–2807.
- Shirogane, T.; Jin, J.; Ang, X.L.; Harper, J.W. SCF -TRCP controls clock-dependent transcription via casein kinase 1-dependent degradation of the mammalian period-1 (Per1) protein. J. Biol. Chem. 2005, 280, 26863–26872.
- Lamia, K.A.; Sachdeva, U.M.; Di Tacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009, 326, 437–440.
- Preitner, N.; Damiola, F.; Molina, L.-L.; Zakany, J.; Duboule, D.; Albrecht, U.; Schibler, U. The Orphan Nuclear Receptor REV-ERBα Controls Circadian Transcription within the Positive Limb of the Mammalian Circadian Oscillator. Cell 2002, 110, 251–260.
- Sato, T.K.; Panda, S.; Miraglia, L.J.; Reyes, T.M.; Rudic, D.; McNamara, P.; Naik, K.A.; FitzGerald, G.A.; Kay, S.A.; Hogenesch, J.B. A Functional Genomics Strategy Reveals Rora as a Component of the Mammalian Circadian Clock. Neuron 2004, 43, 527–537.
- Mitsui, S.; Yamaguchi, S.; Matsuo, T.; Ishida, Y.; Okamura, H. Antagonistic role of E4BP4 and PAR proteins in the circadian oscillatory mechanism. Genes Dev. 2001, 15, 995–1006.
- Eide, E.J.; Vielhaber, E.L.; Hinz, W.A.; Virshup, D.M. The Circadian Regulatory Proteins BMAL1 and Cryptochromes Are Substrates of Casein Kinase Iε. J. Biol. Chem. 2002, 277, 17248–17254.
- Sanada, K.; Okano, T.; Fukada, Y. Mitogen-activated Protein Kinase Phosphorylates and Negatively Regulates Basic Helix-Loop-Helix-PAS Transcription Factor BMAL1. J. Biol. Chem. 2002, 277, 267–271.
- Robles, M.; Humphrey, S.; Mann, M. Phosphorylation Is a Central Mechanism for Circadian Control of Metabolism and Physiology. Cell Metab. 2017, 25, 118–127.
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; HogenEsch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224.
- Panda, S. Circadian physiology of metabolism. Science 2016, 354, 1008–1015.
- Ferrell, J.; Chiang, J.Y. Circadian rhythms in liver metabolism and disease. Acta Pharm. Sin. B 2015, 5, 113–122.
- Stokkan, K.-A.; Yamazaki, S.; Tei, H.; Sakaki, Y.; Menaker, M. Entrainment of the Circadian Clock in the Liver by Feeding. Science 2001, 291, 490–493.
- Eckel-Mahan, K.; Patel, V.R.; De Mateo, S.; Orozco-Solis, R.; Ceglia, N.J.; Sahar, S.; Dilag-Penilla, S.A.; Dyar, K.; Baldi, P.; Sassone-Corsi, P. Reprogramming of the Circadian Clock by Nutritional Challenge. Cell 2013, 155, 1464–1478.
- Vollmers, C.; Gill, S.; DiTacchio, L.; Pulivarthy, S.R.; Le, H.D.; Panda, S. Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 21453–21458.
- Crespo, M.; Gonzalez-Teran, B.; Nikolic, I.; Mora, A.; Folgueira, C.; Rodríguez, E.; Leiva-Vega, L.; Pintor-Chocano, A.; Fernández-Chacón, M.; Ruiz-Garrido, I.; et al. Neutrophil infiltration regulates clock-gene expression to organize daily hepatic metabolism. eLife 2020, 9, 9.
- Watanabe, M.; Tuccinardi, D.; Ernesti, I.; Basciani, S.; Mariani, S.; Genco, A.; Manfrini, S.; Lubrano, C.; Gnessi, L. Scientific evidence underlying contraindications to the ketogenic diet: An update. Obes. Rev. 2020, 21.
- Tognini, P.; Murakami, M.; Liu, Y.; Eckel-Mahan, K.; Newman, J.C.; Verdin, E.; Baldi, P.; Sassone-Corsi, P. Distinct Circadian Signatures in Liver and Gut Clocks Revealed by Ketogenic Diet. Cell Metab. 2017, 26, 523–538.e5.
- Lamia, K.A.; Storch, K.-F.; Weitz, C.J. Physiological significance of a peripheral tissue circadian clock. Proc. Natl. Acad. Sci. USA 2008, 105, 15172–15177.
- Zhang, E.; Liu, Y.; Dentin, R.; Pongsawakul, P.Y.; Liu, A.C.; Hirota, T.; Nusinow, D.; Sun, X.; Landais, S.; Kodama, Y.; et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 2010, 16, 1152–1156.
- Koronowski, K.; Kinouchi, K.; Welz, P.-S.; Smith, J.; Zinna, V.M.; Shi, J.; Samad, M.; Chen, S.; Magnan, C.N.; Kinchen, J.; et al. Defining the Independence of the Liver Circadian Clock. Cell 2019, 177, 1448–1462.e14.
- Sinturel, F.; Gos, P.; Petrenko, V.; Hagedorn, C.; Kreppel, F.; Storch, K.-F.; Knutti, D.; Liani, A.; Weitz, C.; Emmenegger, Y.; et al. Circadian hepatocyte clocks keep synchrony in the absence of a master pacemaker in the suprachiasmatic nucleus or other extrahepatic clocks. Genes Dev. 2021, 35, 329–334.
- Ray, S.; Valekunja, U.K.; Stangherlin, A.; Howell, S.A.; Snijders, A.P.; Damodaran, G.; Reddy, A.B. Circadian rhythms in the absence of the clock gene Bmal1. Science 2020, 367, 800–806.
- Everett, L.J.; Lazar, M.A. Nuclear receptor Rev-erbα: Up, down, and all around. Trends Endocrinol. Metab. 2014, 25, 586–592.
- Guan, D.; Xiong, Y.; Trinh, T.M.; Xiao, Y.; Hu, W.; Jiang, C.; Dierickx, P.; Jang, C.; Rabinowitz, J.D.; Lazar, M.A. The hepatocyte clock and feeding control chronophysiology of multiple liver cell types. Science 2020, 369, eaba8984.
- Reinke, H.; Asher, G. Circadian Clock Control of Liver Metabolic Functions. Gastroenterol. 2016, 150, 574–580.
- Robles, M.; Cox, J.; Mann, M. In-Vivo Quantitative Proteomics Reveals a Key Contribution of Post-Transcriptional Mechanisms to the Circadian Regulation of Liver Metabolism. PLoS Genet. 2014, 10, e1004047.
- Hirayama, J.; Sahar, S.; Grimaldi, B.; Tamaru, T.; Takamatsu, K.; Nakahata, Y.; Sassone-Corsi, P. CLOCK-mediated acetylation of BMAL1 controls circadian function. Nat. Cell Biol. 2007, 450, 1086–1090.
- Edwards, P.A.; Muroya, H.; Gould, R.G. In vivo demonstration of the circadian thythm of cholesterol biosynthesis in the liver and intestine of the rat. J. Lipid Res. 1972, 13, 396–401.
- Yamada, M.; Nagatomo, J.; Setoguchi, Y.; Kuroki, N.; Higashi, S.; Setoguchi, T. Circadian Rhythms of Sterol 12α-Hydroxylase, Cholesterol 7α-Hydroxylase and DBP Involved in Rat Cholesterol Catabolism. Biol. Chem. 2000, 381, 1149–1153.
- Gälman, C.; Angelin, B.; Rudling, M. Bile Acid Synthesis in Humans Has a Rapid Diurnal Variation That is Asynchronous with Cholesterol Synthesis. Gastroenterology 2005, 129, 1445–1453.
- Duez, H.; Van Der Veen, J.N.; Duhem, C.; Pourcet, B.; Touvier, T.; Fontaine, C.; Derudas, B.; Baugé, E.; Havinga, R.; Bloks, V.W.; et al. Regulation of Bile Acid Synthesis by the Nuclear Receptor Rev-erbα. Gastroenterology 2008, 135, 689–698.e5.
- Manieri, E.; Folgueira, C.; Rodríguez, M.E.; Leiva-Vega, L.; Esteban-Lafuente, L.; Chen, C.; Cubero, F.J.; Barrett, T.; Cavanagh-Kyros, J.; Seruggia, D.; et al. JNK-mediated disruption of bile acid homeostasis promotes intrahepatic cholangiocarcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 16492–16499.
- Ma, K.; Xiao, R.; Tseng, H.-T.; Shan, L.; Fu, L.; Moore, D.D. Circadian Dysregulation Disrupts Bile Acid Homeostasis. PLoS ONE 2009, 4, e6843.
- Ferrell, J.; Chiang, J.Y. Short-Term Circadian Disruption Impairs Bile Acid and Lipid Homeostasis in Mice. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 664–677.
- Krishnaiah, S.Y.; Wu, G.; Altman, B.; Growe, J.; Rhoades, S.D.; Coldren, F.; Venkataraman, A.; Olarerin-George, A.O.; Francey, L.J.; Mukherjee, S.; et al. Clock Regulation of Metabolites Reveals Coupling between Transcription and Metabolism. Cell Metab. 2017, 25, 961–974.e4.
- Zhong, X.; Yu, J.; Frazier, K.; Weng, X.; Li, Y.; Cham, C.M.; Dolan, K.; Zhu, X.; Hubert, N.; Tao, Y.; et al. Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m6A mRNA Methylation. Cell Rep. 2018, 25, 1816–1828.e4.
- Solt, L.A.; Wang, Y.; Banerjee, S.; Hughes, T.; Kojetin, D.J.; Lundasen, T.; Shin, Y.; Liu, J.; Cameron, M.D.; Noel, R.; et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nat. Cell Biol. 2012, 485, 62–68.
- Tong, X.; Muchnik, M.; Chen, Z.; Patel, M.; Wu, N.; Joshi, S.; Rui, L.; Lazar, M.A.; Yin, L. Transcriptional Repressor E4-binding Protein 4 (E4BP4) Regulates Metabolic Hormone Fibroblast Growth Factor 21 (FGF21) during Circadian Cycles and Feeding. J. Biol. Chem. 2010, 285, 36401–36409.
- Gavrila, A.; Peng, C.-K.; Chan, J.L.; Mietus, J.E.; Goldberger, A.L.; Mantzoros, C.S. Diurnal and Ultradian Dynamics of Serum Adiponectin in Healthy Men: Comparison with Leptin, Circulating Soluble Leptin Receptor, and Cortisol Patterns. J. Clin. Endocrinol. Metab. 2003, 88, 2838–2843.
- Bass, J.; Takahashi, J. Circadian Integration of Metabolism and Energetics. Science 2010, 330, 1349–1354.
- Kim, C.-W.; Yun, K.E.; Jung, H.-S.; Chang, Y.; Choi, E.-S.; Kwon, M.-J.; Lee, E.-H.; Woo, E.J.; Kim, N.H.; Shin, H.; et al. Sleep duration and quality in relation to non-alcoholic fatty liver disease in middle-aged workers and their spouses. J. Hepatol. 2013, 59, 351–357.
- Kettner, N.; Voicu, H.; Finegold, M.J.; Coarfa, C.; Sreekumar, A.; Putluri, N.; Katchy, C.A.; Lee, C.; Moore, D.D.; Fu, L. Circadian Homeostasis of Liver Metabolism Suppresses Hepatocarcinogenesis. Cancer Cell 2016, 30, 909–924.
- Filipski, E.; Lévi, F. Circadian Disruption in Experimental Cancer Processes. Integr. Cancer Ther. 2009, 8, 298–302.
- Filipski, E.; Subramanian, P.; Carrière, J.; Guettier, C.; Barbason, H.; Lévi, F. Circadian disruption accelerates liver carcinogenesis in mice. Mutat. Res. Toxicol. Environ. Mutagen. 2009, 680, 95–105.
- Mteyrek, A.; Filipski, E.; Guettier, C.; Okyar, A.; Lévi, F. Clock gene Per2 as a controller of liver carcinogenesis. Oncotarget 2016, 7, 85832–85847.
- Fleet, T.; Stashi, E.; Zhu, B.; Rajapakshe, K.; Marcelo, K.L.; Kettner, N.M.; Gorman, B.K.; Coarfa, C.; Fu, L.; O’Malley, B.W.; et al. Genetic and Environmental Models of Circadian Disruption Link SRC-2 Function to Hepatic Pathology. J. Biol. Rhythm. 2016, 31, 443–460.
- Mteyrek, A.; Filipski, E.; Guettier, C.; Oklejewicz, M.; Van Der Horst, G.T.; Okyar, A.; Lévi, F. Critical cholangiocarcinogenesis control by cryptochrome clock genes. Int. J. Cancer 2017, 140, 2473–2483.
- Anafi, R.C.; Francey, L.J.; Hogenesch, J.B.; Kim, J. CYCLOPS reveals human transcriptional rhythms in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, 5312–5317.
- Yang, S.-L.; Yu, C.; Jiang, J.-X.; Liu, L.-P.; Fang, X.; Wu, C. Hepatitis B virus X protein disrupts the balance of the expression of circadian rhythm genes in hepatocellular carcinoma. Oncol. Lett. 2014, 8, 2715–2720.
- Lin, Y.; Chang, J.H.; Yeh, K.; Yang, M.; Liu, T.; Lin, S.; Su, W.; Chang, J. Disturbance of circadian gene expression in hepatocellular carcinoma. Mol. Carcinog. 2008, 47, 925–933.
- Pan, X.; Queiroz, J.; Hussain, M.M. Nonalcoholic fatty liver disease in CLOCK mutant mice. J. Clin. Investig. 2020, 130, 4282–4300.
- Wang, Y.; Lv, K.; Zhao, M.; Chen, H.; Ji, G.; Zhang, Y.; Wang, T.; Cao, H.; Li, Y.; Qu, L. Analysis of miRNA expression profiles in the liver ofClockΔ19mutant mice. PeerJ 2019, 7, e8119.
- Yuan, P.; Li, J.; Zhou, F.; Huang, Q.; Zhang, J.; Guo, X.; Lyu, Z.; Zhang, H.; Xing, J. NPAS2 promotes cell survival of hepatocellular carcinoma by transactivating CDC25A. Cell Death Dis. 2017, 8, e2704.
- Fekry, B.; Ribas-Latre, A.; Baumgartner, C.; Deans, J.R.; Kwok, C.; Patel, P.; Fu, L.; Berdeaux, R.; Sun, K.; Kolonin, M.G.; et al. Incompatibility of the circadian protein BMAL1 and HNF4α in hepatocellular carcinoma. Nat. Commun. 2018, 9, 1–17.
- Manieri, E.; Herrera-Melle, L.; Mora, A.; Tomás-Loba, A.; Leiva-Vega, L.; Fernández, D.I.; Rodríguez, E.; Morán, L.; Cosido, L.H.; Torres, J.L.; et al. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J. Exp. Med. 2019, 216, 1108–1119.
- Singhal, G.; Kumar, G.; Chan, S.; Fisher, F.M.; Ma, Y.; Vardeh, H.G.; Nasser, I.A.; Flier, J.S.; Maratos-Flier, E. Deficiency of fibroblast growth factor 21 (FGF21) promotes hepatocellular carcinoma (HCC) in mice on a long term obesogenic diet. Mol. Metab. 2018, 13, 56–66.