Pancreatic ductal adenocarcinoma (PDA) has long been thought of as a disease arising and progressing from genetic mutations. More recently, it has become clear that epigenetic alterations are also important contributors to PDA progression and metastasis. While no single epigenetic regulator is commonly mutated in PDA, a growing body of evidence indicated that metastatic tissues exhibit distinct epigenetic status compared to primary tumor tissues, which might be exploited for cancer therapeutics and diagnostics.
- pancreatic cancer
- epigenetics
- metastasis
- DNA methylation
1. Introduction
Pancreatic cancer is the third leading cause of cancer-related deaths in the United States. These poor survival outcomes are primarily because pancreatic cancer is often asymptomatic in its early stages, making early diagnoses difficult. The five-year survival rate for pancreatic cancer is 10%, the lowest among common cancers, and pancreatic cancer is expected to surpass colorectal cancer as the second leading cause of cancer-related deaths by 2030 [1][2]. Of pancreatic cancer types, pancreatic ductal adenocarcinoma (PDA) is the most common and aggressive, comprising 90% of pancreatic cancer patients [3]. Despite the fact that recent advances in first-line chemotherapy, such as FOLFIRINOX or gemcitabine/nab-paclitaxel, survival benefits for PDA patients remain modest [4][5]. As a consequence, significant effort has been made to understand the progression of the disease.
Whole-genome sequencing technologies have undoubtedly revealed that PDA is a disease that arises from genetic aberrations. Notably, the initiating genetic event in over 90% of PDA cases is a gain-of-function mutation of KRAS in acinar or ductal cells, which results in the formation of pancreatic lesions called pancreatic intraepithelial neoplasia (PanIN). Subsequent loss-of-function mutations or deletions in tumor suppressor genes, such as TP53, SMAD4 , and CDKN2A , cooperate with KRAS mutation to drive tumor formation and further exacerbate the disease progression ( Figure 1 ) [6][7]. In addition to genetic aberrations, it has become increasingly clear over the last two decades that epigenetic alterations also promote the progression of almost every type of cancer [8][9][10][11]. Epigenetic mechanisms regulate gene transcription, and the proper functioning of these mechanisms is essential for normal development and tissue differentiation. When these mechanisms are aberrantly altered in cancer cells, they can silence tumor suppressor genes or promote the expression of oncogenes to confer advantageous adaptations of the cancer cells, such as increased survival and proliferation, leading to aggressive cell phenotypes and metastasis.
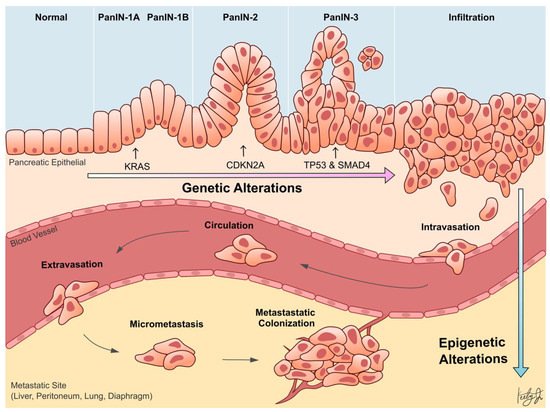
This entry seeks to comprehensively assess the current progress regarding the role of epigenetic alterations in PDA progression and metastasis. Specifically, recent studies investigating the role of alterations in epigenetic regulators, histone modifications, chromatin accessibilities, and DNA methylation in PDA are highlighted ( Table 1 ).
| Category | Gene | Molecular Function | Molecular Phenotype in PDA | Functional Phenotype in PDA | Reference |
|---|---|---|---|---|---|
| Germline PTV Mutation in Epigenetic Regulators |
TET2 | Dioxygenase of 5-methylcytosine, involved in demethylation of cytosines | loss of function in encoded protein | ↓ patient survival | [12] |
| DNMT3a | DNA methyltransferase, involved in de novo DNA methylation | loss of function in encoded protein | ↓ patient survival | [12] | |
| ASXL1 | Polycomb group protein, involved in gene transcriptional regulation and chromatin architecture maintenance | loss of function in encoded protein | ↑ proliferation, ↓ patient survival | [12] | |
| Somatic Mutation in Epigenetic Regulators |
ARID1A | Chromatin remodeler, involved in chromatin remodeling and gene transcriptional regulation | loss of function in encoded protein | ↑ progression, ↓ survival | [6][7][8][13][14] |
| KDM6A | Lysine-specific histone demethylase, involved in promoter and enhancer activities | loss of function in encoded protein | ↑ squamous identity, ↓ survival | [6][7][13][14] | |
| Chromatin Accessibility |
ZKSCAN1 | Transcription factor, involved in proliferation and differentiation | ↑ TF binding via open chromatin | ↑ metastasis | [15] |
| HNF1B | Transcription factor, involved in beta cell development in the pancreas | ↓ TF binding via closed chromatin | ↑ metastasis | [15] | |
| Transcription Factor-Mediated Histone Modification |
FOXA1 | Transcription factor, involved in cell differentiation and chromatin remodeling | ↑ enhancer activation (H3K27ac) | ↑ cell growth, ↑ invasion, ↑ progression | [16] |
| TP63 | Transcription factor, involved in cell proliferation, differentiation, and apoptosis | ↑ enhancer activation (H3K27ac) | ↑ squamous identity | [17] | |
| DNA Methylation | TFPI-2 | Serine proteinase inhibitor, involved in negative regulation of pro-metastasis extracellular matrix degradation | ↓ expression via hypermethylation | ↑ progression, ↑ proliferation, ↑ migration | [18] |
| RELN | Extracellular matrix serine protease, involved in neuronal migration | ↓ expression via hypermethylation | ↓ patient survival, ↑ migration, ↑ invasion, ↑ colony formation | [19] | |
| MET | Receptor tyrosine kinase, involved in cell survival, migration, and invasion | ↑ expression via hypomethylation | ↓ patient survival | [20] | |
| ITGA2 | Integrin, involved in adhesion of cells to the extracellular matrix | ↑ expression via hypomethylation | ↓ patient survival | [20] |
2. Genetic Alterations in Epigenetic Regulators
While KRAS, TP53, SMAD4, and CDKN2A driver mutations are core to early PDA progression, there is a vast genetic heterogeneity among PDA patients, harboring a range of less frequent genetic mutations that facilitate carcinogenesis [6][7]. For one, around 10% of PDA cases belong to familial pancreatic cancer and are commonly affected by germline pre-mature truncating variant (PTV) mutations in genes related to the DNA repair pathways (e.g., BRCA1/2, ATM, and PALB2 ), which have been predicted to inactivate the proteins [6][12]. Interestingly, a subset of these patients also have germline PTV mutations in epigenetic regulators (e.g., TET2 , DNMT3A, and ASXL1 ) [12], suggesting that aberrant changes to the epigenome are important in predisposing individuals to PDA by altering the transcriptional profile of cells.
In addition to the germline mutations in PDA, whole exome and genome sequencing revealed that a significant percentage of patients with PDA have somatic mutations in epigenetic regulators and chromatin remodeling complexes (e.g., ARID1A/B , PBRM1 , MLL2/3/4 , KDM6A , SMARCA2/4 ) [6][7]. Furthermore, somatic mutations in SWI/SNF complex regulators (e.g., ARID1A ) and inactivation of histone modification enzymes (e.g., MLL3 , MLL5 , KDM6A ) frequently occurred in conjunction with oncogenic KRAS in sleeping beauty transposon insertional mutagenesis screens [13][14]. Indeed, Mann et al. found that 100% of the tumors in this screen harbored one or more mutations in genes coding for histone-modifying enzyme [13]. These mutations cooperated with oncogenic KRAS to promote PDA progression, suggesting that alterations to the epigenome are important for driving PDA progression [13]. Together these findings highlight the significance of epigenetic regulation in pancreatic cancer progression.
Despite our firm understanding of the genetic alterations that underlie PDA progression, there is limited understanding of the genetic drivers of PDA metastasis. To this day, there is no known recurrent mutation that drives this metastatic process [15][16][17][18]. This suggests that epigenetic mechanisms, rather than genetic, are driving PDA metastasis ( Figure 1 ). Perturbed epigenetic programs, including transcription factor (TF)-mediated histone modifications, chromatin remodeling , DNA methylation patterns, and subsequently altered transcriptional programs, are emerging mechanisms of PDA progression and metastasis [19].
3. Transcription Factor-Mediated Histone Modification and Metastasis
Several histone post-translational modifications (PTMs) co-occur during chromatin remodeling and gene transcription, which can be used as indicators of transcriptional activities. In general, active promoters are marked by dual H3K27 acetylation (H3K27ac) and H3K4 trimethylation (H3K4me3), while active enhancers are marked by dual H3K27ac and H3K4 monomethylation (H3K4me1) [20]. Conversely, histone H3K9 and H3K27 methylation are used to indicate repressive gene transcription [21]. These histone PTMs alter biochemical properties of the chromatin, not only leading to the formation of euchromatin or heterochromatin, but also the sequestering or docking of effector enzymes, such as histone acetylase, deacetylase, methyltransferase, and demethylase [22].
Dysregulation in histone PTMs, in conjunction with the recruitment of effector chromatin remodelers, modifies chromatin architecture, leading to aberrant gene activation or repression, which contributes to cancer metastasis ( Figure 2 ). For example, by comparing primary PDA tumors to matched distant lung and proximal peritoneum metastases, McDonald et al. used chromatin immunoprecipitation followed by sequencing (ChIP-seq) to demonstrate that global alterations of H3K9me2/3 and H3K27ac may contribute to aggressive tumor phenotypes [23]. Specifically, ChIP-seq showed that H3K9 methylation levels are reduced at Large Organized Chromatin K9-modified (LOCK) heterochromatin regions in distant metastases compared to their matched primary tumors [23], suggesting that transcription activities of certain genes within these regions (LOCK genes) are upregulated during PDA metastasis. Indeed, using RNA-seq and ChIP-seq analysis, the study showed that decreased H3K9me2 and H3K27me3 and increased H3K27ac occupancies at gene promoters in LOCK regions is positively correlated with expression of the associated genes in distant metastases [23]. Furthermore, subsequent gene ontology analysis revealed that the reprogrammed LOCK regions contain genes related to cellular differentiation and morphogenesis, epithelial-to-mesenchymal transitions, cell adhesion, and migration [23]. This suggests that a histone modification-mediated epigenetic switch from heterochromatin to euchromatin state is associated with cellular transformation, which promotes aggressive tumor phenotypes and facilitates PDA tumor-to-metastasis transitions.
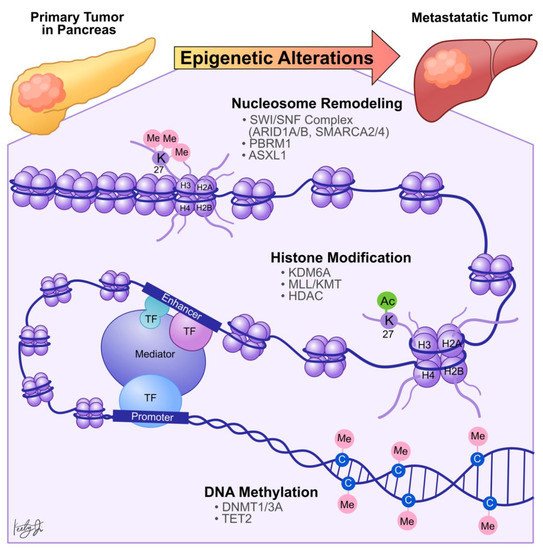
Histone modification-dependent epigenetic landscape reprogramming can be carried out by TFs through first targeting nucleosomal DNA and then recruiting histone and chromatin remodeling enzymes [24]. In the context of pancreatic cancer, we identified that the developmental Forkhead family TF FOXA1 drives enhancer landscape reprogramming during PDA tumor-to-metastasis transition [25]. To dissect the molecular mechanisms of enhancer activation/inactivation during PDA metastasis, we developed 3D organoid culture using PDA cells collected from the primary tumors and matched metastatic lesions derived from the Kras +/LSL-G12D ; Trp53 +/LSL-R172H ; Pdx1-Cre (KPC) PDA mouse model [26]. The organoid culture model of PDA preserves the unique biological characteristics of normal, PanIN, tumor, and metastases lesions. In addition, this model can be used for direct biochemical comparisons during each stage of the disease progression [26]. H3K27ac ChIP-seq analysis revealed 857 regions with increased H3K27ac occupancy ( GAIN region) in the metastases organoids compared to the normal, PanIN, and tumor organoids [25], suggesting that the dysregulation of H3K27ac landscape within these enhancer regions could be responsible for PDA progression and metastasis. Combining RNA-seq and TF motif analysis, we then identified that FOXA1 activates GAIN region enhancers by increasing H3K27ac occupancy in the primary PDA. In vitro , overexpression of FOXA1 in primary PDA tumor cells activated foregut developmental genes that promoted anchorage-independent cell growth and invasion in sphere-formation and Matrigel invasion assays, respectively. In vivo , overexpression of FOXA1 contributed to overall PDA progression and metastasis in tail vein injection and organoid transplantation experiments [25]. This work demonstrated that PDA cells could repurpose FOXA1 to activate enhancers of developmental gene programs [27], promote anchorage-independent growth, and induce branching morphogenesis of the epithelial cells [28][29]. Furthermore, upregulation of FOXA1 in PDA cells promotes aggressive cell phenotypes, such as proliferation, invasion, and migration, allowing cells to better withstand stressful conditions during metastasis. In support of our work, Kim et al. discovered that FOXA1 gene transcription is enhanced by missense mutations of p53 (p53 R172H , p53 R245W , and p53 R270H ) that directly bind to the FOXA1 promoter and induce oncogenic KRAS activation of cyclic AMP responsive element binding protein 1 (CREB1) [30]. In turn, FOXA1 promotes β-catenin stabilization and subsequently activates canonical WNT transcriptional programs to promote anchorage-independent cell growth, proliferation, and metastasis [30][31]. Together, these studies demonstrated that PDA cells could reprogram the epigenetic landscape and subsequent transcription programs through (1) recruiting TFs, (2) altering chromatin architectures through histone modifications, and (3) recruiting transcription co-activators (i.e., mutant p53 and CREB1) to sustain their growth, differentiation, and metastasis ( Figure 2 ).
4. DNA Methylation and Metastasis
In PDA, aberrant DNA methylation has been widely documented. Early on, these studies involved methylated CpG island amplification (MCA) followed by microarray sequencing. More recently, bisulfite treatment paired with large microarray platforms or next-generation sequencing, such as reduced-representative bisulfite sequencing (RRBS) or whole-genome bisulfite sequencing (WGBS), have been used to assess the DNA methylome at base-pair resolution. Using these methods, DNA methylation in PDA has been correlated with several disease outcomes and histopathological phenotypes. For example, Thompson et al. identified 17,251 CpG sites that are negatively associated with survival outcome and 3256 sites that are positively associated with survival outcome in a comparison of RRBS data from a small cohort of PDA patient tissues and adjacent normal pancreas tissues [32]. Similarly, Mishra et al. identified 406 promoter methylation loci associated with survival in an analysis of 450K array methylation data from the 154 PDA samples in The Cancer Genome Atlas pancreatic cancer patient database (TCGA-PAAD) [33]. Unsupervised clustering of TCGA-PAAD samples based on the differentially methylated CpG sites resulted in three distinct clusters of patient samples [34]. These clusters were each enriched with a different tumor grade, indicating that DNA methylation can be used to estimate the histopathological stage of PDA tumors [34]. In an analysis of both TCGA-PAAD transcriptome and DNA methylome data, unsupervised subtyping of TCGA-PAAD samples based on genes whose expression was significantly correlated with methylation expression patterns was performed [35]. Interestingly, this analysis identified five subtypes, four of which correspond to the molecular subtypes identified by Bailey et al. (i.e., squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine [ADEX]), and the last unique subtype was enriched for tumor microenvironment related genes [36][35]. Together, these data suggest that aberrant DNA methylation is associated with aggressive PDA phenotypes.
To identify pathways that may be involved in DNA methylation-mediated PDA aggressiveness, gene ontology and/or pathway analysis is often performed on differentially methylated genes in PDA. In the Thompson et al. study, CpG sites with a negative correlation between methylation and survival rate were associated with pancreas-specific development genes [32]. Normally, pancreas-specific development genes are only active in early embryonic stages, but reactivation of these genes during PDA is common [37][38]. Differential methylation of pancreas development genes has also been noted in the TCGA-PAAD dataset, suggesting that reactivation of the embryonic pancreas development program in PDA is epigenetically regulated [34]. Other differentially methylated genes found in the TCGA-PAAD dataset were enriched for cancer-related pathways, including MAPK, Rap1, and calcium signaling [33][34]. In addition, core signaling pathways that are commonly altered in PDA, such as Wnt/Notch signaling, apoptosis, cell-cycle regulation, and cell adhesion, were enriched in aberrantly methylated genes found in the analysis of TCGA-PAAD database as well as a separate bisulfite microarray study of 167 PDA and 29 adjacent normal pancreata conducted by Nones et al. [39][34][40]. Interestingly, Nones et al. also observed that stellate cell activation genes were often hypomethylated and therefore, likely downregulated in PDA [39]. In support of this, Espinet et al. discovered that stellate cells exposed to conditioned media derived from high-interferon (IFN) signature patient tissues showed an increased stellate cell growth in vitro and tumor formation in vivo [41]. Because the IFN pathway is involved in anti-viral defense and activated stellate cells are involved in ECM remodeling, this pathway may be activating and reprogramming stellate cells to produce a pro-inflammatory stroma that facilitates tumor growth. Hypermethylation of homeobox genes, which encode key transcription factors of embryonic development, was also commonly detected in several PDA methylome studies [42][34][41][43]. This provides additional evidence that PDA tumors reactivate developmental pathways via epigenetic mechanisms to promote metastatic characteristics. Overall, these studies suggest that processes commonly implicated in cancer aggressiveness and metastasis, such as apoptosis, cell-cycle regulation, and development pathways, are heavily influenced by aberrant DNA methylation in PDA.
Aberrant methylation of several individual genes and their association with worse survival outcomes have also been documented. Most of these genes have been reported to have hypermethylated promoters. For example, Sato et al. showed that the low expression of TFPI-2 , which encodes a negative regulator of pro-metastasis extracellular matrix degradation, is frequently seen in both PDA cell lines and primary tumors and is associated with hypermethylation of the TFPI-2 promoter [44]. Restoration of TFPI-2 in the PDA cell lines reduced proliferation and migratory potential [44]. Likewise, RELN , which encodes a critical regulator of neuronal migration, is commonly hypermethylated and silenced in pancreatic cancer [45]. Furthermore, low expression of RELN was significantly associated with worse survival outcomes, and siRNA knockdown of RELN in RELN-expressing pancreatic cancer cells enhanced cell motility, invasion, and colony formation [45]. Nevertheless, promoter hypomethylation of certain genes has also been implicated in a worse prognosis. For example, hypomethylation of MET , whose aberrant expression promotes metastasis, and ITGA2 , which is involved in cell adhesion, correlated with increased mRNA expression and associated with poor survival in PDA by Nones et al. [39]. Thus, aberrant promoter methylation likely contributes to the aggressive nature of PDA by altering the expression of genes such as TFPI-2 , RELN , MET , and ITGA2 .
While the above studies provide a strong indication that aberrant DNA methylation in PDA likely contributes to metastasis, it is important to note that many of these studies are largely association-based and therefore do not functionally implicate genes or pathways in the process. Studies with functional experiments have shown that altering the expression of aberrantly methylated genes with loss-of-function or gain-of-function approaches affects metastatic potential in vitro , but analogous experiments in vivo are lacking. Furthermore, no study has directly shown the consequences of altered DNA methylation on the metastatic characters in PDA. Mechanistic studies linking aberrant DNA methylation to the aggressive behavior of PDA in both in vitro and in vivo contexts are necessary to better elucidate the role of DNA methylation in PDA metastasis.
This entry is adapted from the peer-reviewed paper 10.3390/biom11081082
References
- Roberts, N.J.; Norris, A.L.; Petersen, G.M.; Bondy, M.L.; Brand, R.; Gallinger, S.; Kurtz, R.C.; Olson, S.H.; Rustgi, A.K.; Schwartz, A.G.; et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016, 6, 166–175.
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13.
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501.
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505.
- Mann, K.M.; Ward, J.M.; Yew, C.C.K.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Chang, D.K.; et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941.
- Pérez-Mancera, P.A.; Rust, A.G.; Van Der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.T.; Grützmann, R.; Aust, D.; Rümmele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270.
- Dhara, S.; Chhangawala, S.; Chintalapudi, H.; Askan, G.; Aveson, V.; Massa, A.L.; Zhang, L.; Torres, D.; Makohon-Moore, A.P.; Lecomte, N.; et al. Pancreatic cancer prognosis is predicted by an ATAC-array technology for assessing chromatin accessibility. Nat. Commun. 2021, 12, 3044.
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20.
- Somerville, T.D.D.; Xu, Y.; Miyabayashi, K.; Tiriac, H.; Cleary, C.R.; Maia-Silva, D.; Milazzo, J.P.; Tuveson, D.A.; Vakoc, C.R. TP63-Mediated Enhancer Reprogramming Drives the Squamous Subtype of Pancreatic Ductal Adenocarcinoma. Cell Rep. 2018, 25, 1741–1755.e7.
- Sato, N.; Parker, A.R.; Fukushima, N.; Miyagi, Y.; Iacobuzio-Donahue, C.A.; Eshleman, J.R.; Goggins, M. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene 2005, 24, 850–858.
- Sato, N.; Fukushima, N.; Chang, R.; Matsubayashi, H.; Goggins, M. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology 2006, 130, 548–565.
- Nones, K.; Waddell, N.; Song, S.; Patch, A.M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P.; et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int. J. Cancer 2014, 135, 1110–1118.
- Mann, K.M.; Ward, J.M.; Yew, C.C.K.; Kovochich, A.; Dawson, D.W.; Black, M.A.; Brett, B.T.; Sheetz, T.E.; Dupuy, A.J.; Chang, D.K.; et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2012, 109, 5934–5941.
- Pérez-Mancera, P.A.; Rust, A.G.; Van Der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.T.; Grützmann, R.; Aust, D.; Rümmele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270.
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; Lin, M.L.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113.
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of Genomic and Transcriptional Features in Pancreatic Cancer Reveals Increased Cell Cycle Progression in Metastases. Cancer Cell 2019, 35, 267–282.e7.
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366.
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216.
- Embuscado, E.E.; Laheru, D.; Ricci, F.; Yun, K.J.; Witzel, S.D.B.; Seigel, A.; Flickinger, K.; Hidalgo, M.; Bova, G.S.; Iacobuzio-Donahue, C.A. Immortalizing the complexity of cancer metastasis: Genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol. Ther. 2005, 4, 548–554.
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, 324.
- Wong, C.M.; Wong, C.C.L.; Ng, Y.L.; Au, S.L.K.; Ko, F.C.F.; Ng, I.O.L. Transcriptional repressive H3K9 and H3K27 methylations contribute to DNMT1-mediated DNA methylation recovery. PLoS ONE 2011, 6, e16702.
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783.
- McDonald, O.G.; Li, X.; Saunders, T.; Tryggvadottir, R.; Mentch, S.J.; Warmoes, M.O.; Word, A.E.; Carrer, A.; Salz, T.H.; Natsume, S.; et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet. 2017, 49, 367–376.
- Mayran, A.; Drouin, J. Pioneer transcription factors shape the epigenetic landscape. J. Biol. Chem. 2018, 293, 13795–13804.
- Roe, J.S.; Hwang, C.I.; Somerville, T.D.D.; Milazzo, J.P.; Lee, E.J.; Da Silva, B.; Maiorino, L.; Tiriac, H.; Young, C.M.; Miyabayashi, K.; et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell 2017, 170, 875–888.e20.
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.C.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338.
- Gao, N.; LeLay, J.; Vatamaniuk, M.Z.; Rieck, S.; Friedman, J.R.; Kaestner, K.H. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008, 22, 3435–3448.
- Vigil, D.; Martin, T.D.; Williams, F.; Jen Yeh, J.; Campbell, S.L.; Der, C.J. Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J. Biol. Chem. 2010, 285, 34729–34740.
- Larsen, H.L.; Grapin-Botton, A. The molecular and morphogenetic basis of pancreas organogenesis. Semin. Cell Dev. Biol. 2017, 66, 51–68.
- Kim, M.P.; Li, X.; Deng, J.; Zhang, Y.; Dai, B.; Allton, K.L.; Hughes, T.G.; Siangco, C.; Augustine, J.J.; Kang, Y.; et al. Oncogenic KRAS recruits an expansive transcriptional network through mutant p53 to drive pancreatic cancer metastasis. Cancer Discov. 2021.
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473.
- Thompson, M.J.; Rubbi, L.; Dawson, D.W.; Donahue, T.R.; Pellegrini, M. Pancreatic Cancer Patient Survival Correlates with DNA Methylation of Pancreas Development Genes. PLoS ONE 2015, 10, e0128814.
- Mishra, N.K.; Southekal, S.; Guda, C. Survival analysis of multi-omics data identifies potential prognostic markers of pancreatic ductal adenocarcinoma. Front. Genet. 2019, 10, 624.
- Mishra, N.K.; Guda, C. Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer. Oncotarget 2017, 8, 28990–29012.
- Roy, S.; Singh, A.P.; Gupta, D. Unsupervised subtyping and methylation landscape of pancreatic ductal adenocarcinoma. Heliyon 2021, 7.
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52.
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249.
- Koizumi, M.; Doi, R.; Toyoda, E.; Masui, T.; Tulachan, S.S.; Kawaguchi, Y.; Fujimoto, K.; Gittes, G.K.; Imamura, M. Increased PDX-1 expression is associated with outcome in patients with pancreatic cancer. Surgery 2003, 134, 260–266.
- Nones, K.; Waddell, N.; Song, S.; Patch, A.M.; Miller, D.; Johns, A.; Wu, J.; Kassahn, K.S.; Wood, D.; Bailey, P.; et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int. J. Cancer 2014, 135, 1110–1118.
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806.
- Espinet, E.; Gu, Z.; Imbusch, C.D.; Giese, N.A.; Buscher, M.; Safavi, M.; Weisenburger, S.; Klein, C.; Vogel, V.; Falcone, M.; et al. Aggressive PDACs show hypomethylation of repetitive elements and the execution of an intrinsic IFN program linked to a ductal cell-of-origin. Cancer Discov. 2020, 11, 638–659.
- Vincent, A.; Omura, N.; Hong, S.M.; Jaffe, A.; Eshleman, J.; Goggins, M. Genome-wide analysis of promoter methylation associated with gene expression profile in pancreatic adenocarcinoma. Clin. Cancer Res. 2011, 17, 4341–4354.
- Tan, A.C.; Jimeno, A.; Lin, S.H.; Wheelhouse, J.; Chan, F.; Solomon, A.; Rajeshkumar, N.V.; Rubio-Viqueira, B.; Hidalgo, M. Characterizing DNA methylation patterns in pancreatic cancer genome. Mol. Oncol. 2009, 3, 425–438.
- Sato, N.; Parker, A.R.; Fukushima, N.; Miyagi, Y.; Iacobuzio-Donahue, C.A.; Eshleman, J.R.; Goggins, M. Epigenetic inactivation of TFPI-2 as a common mechanism associated with growth and invasion of pancreatic ductal adenocarcinoma. Oncogene 2005, 24, 850–858.
- Sato, N.; Fukushima, N.; Chang, R.; Matsubayashi, H.; Goggins, M. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology 2006, 130, 548–565.