Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biology
Perturbed cardiac fetal blood flow and congenital heart disease are intrinsically interlinked. The presence of subtle or overt structural heart defects induces blood flow conditions that differ from normal. Because blood flow provides important mechanical feedback during heart development, mutations in genes that are responsive to flow (such as the vascular endothelial growth factor family) or perturbations in blood flow itself both lead to congenital heart defects. In particular tetralogy of Fallot (TOF) is associated with perturbations in early embryonic blood flow as well as genetic mutations.
- hemodynamics
- cardiac malformations
- flow-induced heart defects
- VEGF
- semaphorin signaling
- cardiac neural crest cells
- outflow tract
1. The Vascular Endothelial Growth Factor (VEGF) Family
The vascular endothelial growth factor (VEGF) pathway is one of the most critical pathways for vascular development. In brief, secreted VEGF ligands bind to transmembrane VEGF receptors, which heterodimerize or homodimerize and autophosphorylate upon ligand binding (see Figure 5). Among the VEGF proteins, VEGF-A is the major family member associated with blood vessel formation and can generate numerous isoforms via alternative splicing. Among the 16 isoforms, VEGF-A165 was the first identified and remains the prototypical pro-angiogenic isoform (reviewed in [33]). VEGF receptor 2 (VEGFR2) can further signal independent of VEGF-A in the presence of high wall shear stress [34]. Adding more complexity to this pathway, VEGF receptors may heterodimerize with co-receptor neuropilin-1, which can also form heterodimers with the plexin receptors of the semaphorin-plexin signaling axis (Figure 5). Neuropilin-1 thus serves as a molecular switch between pro-angiogenic VEGF-A signaling and anti-angiogenic semaphorin signaling. Because vascular development relies on the level of VEGF-A signaling, mutations in any component of the VEGF-A or semaphorin pathways may lead to phenotypic anomalies. We will focus on mutations within these pathways that lead to tetralogy of Fallot (TOF) versus persistent truncus arteriosus heart defects and/or are associated with perturbations in blood flow.
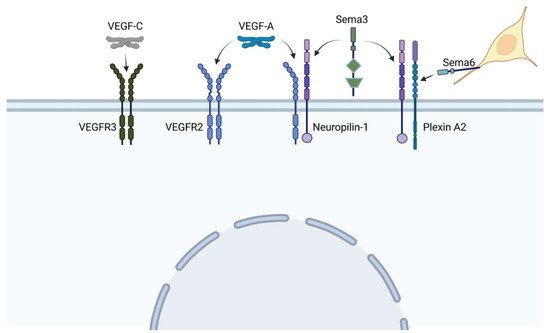
Figure 5. VEGF and sempahorin (Sema) ligands bind to a variety of heterodimer receptors, and co-receptor Neuropilin-1 can heterodimerize with both VEGF and plexin receptors, leading to extensive cross-talk between pathways. Neural crest cells (yellow) express the membrane-bound semaphorin 6, which directs their migration toward specific plexin receptors. (Created with BioRender).
2. Mutations within the VEGF-A Pathway Are Associated with TOF
Patients with TOF and mutations in the VEGF pathway have an increased risk of presenting with an absent pulmonary valve and a right-sided aortic arch compared with patients with TOF who have other genetic variations [35]. Single nucleotide polymorphisms (SNPs) within VEGF-A that have been associated with TOF include -C2578A (rs699947), -C634G (rs2010963), and C936T (rs3025039). These specific SNPs are within the promoter or untranslated regions of the VEGF-A gene and reduce VEGF-A expression, thus increasing the risk of developing TOF [36]. Intriguingly, an estrogen receptor within the VEGF-A promoter limits the effects of the C936T substitution among females and reduces their risk of developing TOF [37,38].
Genetic manipulation of the VEGF-A expression level in mouse supports the importance of VEGF-A during development. When the first VEGF-A knockout mice were generated, even lacking one copy of this gene resulted in mid-gestational lethality due to abnormal yolk sac vasculogenesis, impaired hematopoiesis, and stunted heart development [39,40]. A hypomorphic lacZ-knock-in VEGF-A allele allowed for the survival of fertile heterozygous mice, but the homozygous hypomorphs still died by embryonic day 9.0 and show the same impaired hematopoiesis and vasculogenesis as observed in the original VEGF-A heterozygotes [41].
VEGF receptor 2 (VEGFR2, also known as KDR) is broadly expressed in the vascular endothelium and blood islands and is critical for their development [21,22,23,24]. The global VEGFR2 knockout is embryonic lethal between embryonic days 8.5 and 9.5 due to failure to form the blood islands or blood vessels [42]. At later stages, this receptor is maintained in the endothelium of the pharyngeal arch arteries, where it is phosphorylated in response to blood flow. Although both the left and right sixth PAAs express VEGFR2, reduced blood flow through the right sixth arch artery means that VEGF signaling is absent; thus, only the left sixth PAA, with its active VEGF signaling, is maintained and remodeled to give rise to the ductus arteriosus [43]. Although PAAs III and IV express VEGF receptor 2 [43], they do not regress if VEGFR2 phosphorylation is inhibited [44]. This difference may be related to differential neural patterning through the arches, though the molecular mechanisms remain unclear (reviewed in [45]). Stop-gain and deleterious missense mutations in VEGFR2 have been identified in patients with TOF, particularly those with absent pulmonary valve or complete pulmonary atresia [35]. These phenotypes are consistent with VEGFR2’s critical role in maintaining the left sixth PAA.
Unexpectedly, mutations within VEGF receptor FLT4 (i.e., VEGFR3) can also lead to TOF. VEGFR3 is initially expressed in small mesenchymal clusters within the pharyngeal arches (embryonic day 9.5–10.5) and becomes restricted to Prox1-positive lymphatic cells during outflow tract septation [46]. VEGFR3 is typically associated with lymphangiogenesis because it promotes lymphatic endothelial cell migration upon binding to VEGF-C [47]. This receptor is indispensable for lymphatic vessels, and mutations are associated with lymphatic disorders. However, loss-of-function stop codons and nonsense mutations are associated with TOF and are distinct from the mutations that lead to lymphatic disorders [48]. VEGFR3′s restricted expression pattern, particularly because it is not expressed in the outflow tract endothelium [46], makes it challenging to visualize how truncation mutations specifically lead to TOF. Additionally, knockout mouse models exhibit cardiac failure and embryonic lethality by embryonic day 10.5 [49], preventing an analysis of how loss of VEGFR3 relates to the specific development of TOF.
3. Mutations within the VEGF-Semaphorin Crosstalk Mediator Neuropilin-1 Can Lead to TOF or Persistent Truncus Arteriosus
Among VEGF’s roles, low levels of VEGF promote the endothelial expression of Semaphorin 3A, which then recruits neuropilin-1-expressing monocytes that stabilize developing blood vessels [50]. In contrast, high levels of VEGF inhibit vascular stability but are needed to initiate angiogenesis [50]. These reciprocal inhibitions suggest that the VEGF and semaphorin signaling pathways have distinct, critical roles during angiogenesis. Both ligands can bind to co-receptor neuropilin-1, which thus plays a key role in switching from VEGF to semaphorin signaling. Neuropilin-1 is a single-pass transmembrane protein with five distinct extracellular domains. Two domains (a1 and a2) facilitate semaphorin binding; two other domains (b1 and b2) facilitate VEGF-A165 binding. The final extracellular domain is the MAM domain, which may support—but does not directly cause—dimerization with other receptors [51]. This MAM domain is the major distinction between endothelial-specific, full-length neuropilin-1 and soluble neuropilin-1 isoforms that lack this domain and are not expressed in the endothelium.
Full-length neuropilin-1 enhances VEGF-A165 binding to VEGFR2 and promotes cytoskeletal remodeling [52]. When neuropilin-1 is conditionally knocked out of endothelial cells, the resulting mice exhibit a single outflow vessel. Although neuropilin-1 can bind to both semaphorin and VEGF, the VEGF binding appears key for its role in outflow tract septation. Knocking in a neuropilin-1 mutation that eliminates the semaphorin-binding site yields normal outflow tract septation [53].
Two neuropilin-1 mutations are associated with TOF. V733I occurs within the MAM domain and is predicted to yield a nonsense mutation [54,55]. Neuropilin-1 splice variants that lack the MAM domain lead to soluble isoforms that act as decoy receptors to downregulate VEGF signaling [56]. These soluble neuropilin splice variants specifically sequester VEGF-A165, thus inhibiting VEGF-A’s functions and leading to increased endothelial apoptosis, decreased blood vessel numbers, and decreased vascular density [57,58]. Intriguingly, VEGF-A induces a proteolytic cleavage of the extracellular binding domains (a1–b2) of neuropilin-1, creating an additional soluble neuropilin-1 decoy that downregulates both VEGF and semaphorin signaling, creating a negative feedback loop [52,59].
The other TOF-associated neuropilin-1 mutation is within the 3′ UTR (rs10080) and is predicted to be a miRNA-binding site [55]. A patient presented with TOF with double aortic arch and a 1.8 Mb deletion on chromosome 11 that reduced their expression level of neuropilin-1 [59]. Neuropilin-1 can repress VEGF-A signaling when neuropilin-1 and VEGFR2 are expressed on adjacent cells, forming trans heterodimers [60]; thus, the neuropilin-1 expression is likely key for normal vascular development.
In contrast to the neuropilin-1 variants that lack the MAM domain and supporting neuropilin-1′s role in mediating a switch between VEGF and semaphorin signaling, a splice site mutation that leads to a truncated neuropilin-1 has been linked to persistent truncus arteriosus [61]. In the patient with this truncated neuropilin-1, the truncation prevents the translation of any functional protein. This patient data is supported by the neuropilin-1 knockout mouse, which presents with regression of left PAA IV and VI and results in persistent truncus arteriosus [62].
Adding additional complexity to the neuropilin-VEGF pathway, whether neuropilin-1 and VEGFR2 form cis heterodimers within a single cell or trans heterodimers when expressed in different cells determines which downstream ERK is phosphorylated. Activation of the trans heterodimers specifically phosphorylates the kinase ERK2, compared with cis heterodimers leading to ERK1 phosphorylation [60]. Of these two kinases, ERK2 is located near the classical 22q11.2 deletion, and haploinsufficiency of ERK2 is associated with persistent truncus arteriosus and right-sided aortic arch [63].
4. Mutations within the Semaphorin Signaling Pathway Lead to TOF or Persistent Truncus Arteriosus
The canonical semaphorin signaling pathway consists of the semaphorin ligands, which may be secreted (e.g., class III) or transmembrane (e.g., class VI) proteins, and their plexin receptors, which fall into four major classes (A–D). As with the VEGF pathway, the receptors form heterodimers or homodimers, and heterodimers can be formed with neuropilin 1 [64]. The downstream effects of each pathway depend on the affinity of the semaphorin ligands that are present for the receptors that are expressed, and many of these ligand-receptor combinations impact vascular development.
Among the class III semaphorins, semaphorin 3A is flow-sensitive, and its expression is enriched in the anti-atherosclerotic portion of the adult aortic arch and is downregulated by oscillatory flow [65]. Additionally, it is the semaphorin ligand with the highest binding affinity for neuropilin-1 [56]. When VEGF-A levels decrease, semaphorin 3A helps recruit neuropilin 1-expressing monocytes to stabilize blood vessels [50]. Semaphorin 3A competitively inhibits VEGF-A165 from binding to neuropilin-1-VEGFR2 heterodimers and can block VEGF-A165 and VEGF-A121 activity in a neuropilin-1-indepdendent manner [66]. Thus, mutations in semaphorin 3A may lead to aberrantly high levels of VEGF signaling or the inability to halt VEGF-induced chemotaxis, causing over-remodeling at the expense of vascular stabilization. Perhaps unsurprisingly, semaphorin 3A mutations and copy number variations in semaphorin 3D and 3E are associated specifically with TOF [67]. Vascular instability may help explain the pulmonary stenosis or atresia observed in TOF.
Semaphorin 3C plays a key role in directing cardiac neural crest cells to the outflow tract. It is expressed in the second heart field mesoderm and repressed in the neural crest cells as the second heart field is adding to the outflow tract and the cardiac neural crest cells have reached the pharyngeal arches [68]. As the cardiac neural crest migrate into the outflow tract, semaphorin 3C is then observed in the myocardial cuff surrounding the outflow tract and the cardiac neural crest themselves [69,70]. Because semaphorin 3C causes aggregation of the cardiac neural crest [68], these expression patterns suggest that semaphorin 3C is key for outflow tract septation. Receptors plexin D1 and neuropilin-1 are present in the endothelium of the aortic arch [70]. In the semaphorin 3C-null mouse embryo, the cardiac neural crest fails to migrate into the outflow tract [71], and semaphorin 3C-null mice exhibit persistent truncus arteriosus and interrupted aortic arch types B and C [72].
Unique among the semaphorins, semaphorin 3E can bind to plexin D1 in the presence [73] or absence [74] of neuropilin. Thus, this ligand may serve as another switch between plexin D1-associated persistent truncus arteriosus and plexin A2-associated TOF. When bound to plexin D1, semaphorin 3E inhibits endothelial migration and tube formation by destabilizing the actin cytoskeleton [75]. Mutations in semaphorins 3E and 4A, along with receptor plexin D1, are associated with persistent truncus arteriosus [76,77], which suggests that these guidance cues are critical for neural crest migration during arterial pole septation. However, when semaphorin 3E binds plexin D1, it can block VEGF-induced Akt phosphorylation to inhibit endothelial migration and tube formation [78]. Insufficiency for semaphorin 3E would thus allow excessive endothelial migration, suggesting a mechanism for pulmonary stenosis or atresia. In support of this hypothesis, some patients with TOF have copy number loss of chromosome 7q21.11, which contains semaphorin 3D and semaphorin 3E [67].
Intriguingly, though, plexin D1 is a mechanosensor that can form a complex with neuropilin-1 and VEGFR2 and can act independently of semaphorin 3E to induce a response to wall shear stress [79]. This force-transduction role opens new questions about how plexin D1 and its semaphorin ligands contribute to outflow tract and pharyngeal arch artery remodeling in response to blood flow.
Despite including both class III secreted (3A, 3C, and 3F) and class VI transmembrane (6A and 6B) semaphorin ligands, all five of these ligands can bind to receptor plexin A2 [70,80,81]. Plexin A2 is expressed by cardiac neural crest cells and is observed in the mesenchyme of the pharyngeal arches and the outflow tract during cardiac neural crest migration toward the outflow tract [70,71]. During outflow tract septation, plexin A2 is particularly enriched where the neural crest prongs are fusing together [69]. Cardiac neural crest cells express plexin A2 and are thus attracted to semaphorin 3C, which is observed in the outflow tract myocardium, but repulsed from cells that express transmembrane semaphorin 6A or 6B. Semaphorin 6A and 6B are predominantly expressed in the dorsal neural tube and somites during neural crest migration and may thus help direct the neural crest to migrate ventrally. Consistently, the plexin A2-null mouse displays persistent truncus arteriosus and two variations of interrupted aortic arch [70]. However, these phenotypes show variable penetrance, underscoring the complexity of compensation. Highlighting this complexity, loss of plexin A2 copy number has been identified in patients with TOF [67].
This entry is adapted from the peer-reviewed paper 10.3390/jcdd8080090
This entry is offline, you can click here to edit this entry!