Glioblastoma is the most frequent and the most aggressive brain tumor. Even with the most current treatment, its prognosis remains dismal. Immunotherapies, novel cancer therapies using the patient’s own immune system to fight cancer, have revolutionized the treatment of numerous cancer types and generate great hope for glioblastoma. In this review, we analyze the challenges immunotherapy is facing in glioblastoma, present the different immunotherapy approaches with corresponding key clinical trial findings, and finally discuss limitations and how they might be overcome. Proof of efficacy for immunotherapies remains to be demonstrated in glioblastoma, but novel combinatorial approaches remain promising.
- glioblastoma
- immunotherapy
- checkpoint inhibitor
- vaccine
- oncolytic virus
- CAR T cell
1. Introduction
2. Standard of Care
3. Immune Privilege of the CNS
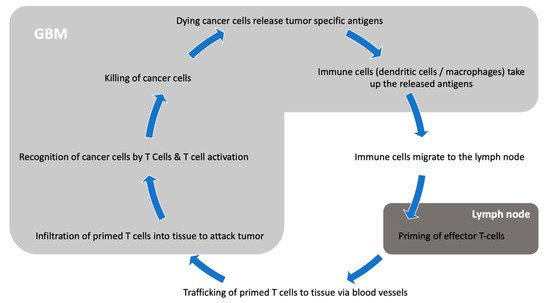
4. Mechanisms of Immune Evasion by GBM
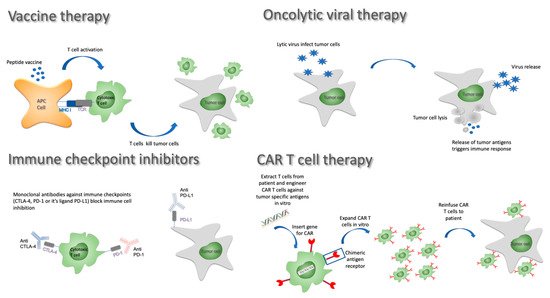
5. Immunotherapy Strategies
5.1. Vaccine Approaches
| Clinical Trial Name | Description | Phase of Trial | Nb of Participants | Primary Outcome Measure | mOS | mPFS | Significant Results | Demonstrated Immune Response |
Comments |
|---|---|---|---|---|---|---|---|---|---|
| Vaccines | |||||||||
| Phase III trials | |||||||||
| NCT01480479 (ACT IV) | Rindopepimut + TMZ in newly diagnosed EGFRvIII positive patients | 3 | 745 | OS | 20.1 mo | 8.0 mo | No difference in OS (2.1 vs. 20.0 mos) and PFS (8.0 vs. 7.4 mos) | Robust systemic antigen-specific antibody response | Subgroup analysis suggests a modest advantage in patients with residual tumors (≥2 cm2) |
| NCT00045968 (DCVax-L) | DCvax-L in newly diagnosed GBM following resection | 3 | 348 | PFS | 23.1 mo | not yet available | So far, only OS result of the combined arms reported | Not reported (yet) | No clear indication of positive effect/1599 patients screened and 348 patients included |
| Randomized trials | |||||||||
| NCT01280552 (ICT-107) | Double-blind, randomized study of ICT-107 with maintenance TMZ in newly diagnosed GBM | 2 | 124 | OS | 17.0 vs. 15.0 mos (HR: 0.87; p = 0.58) | 11.2 vs. 9.0 mos (HR = 0.57, p = 0.58) | No difference in OS, PFS in favor of treatment arm | Robust systemic response | Pts in the HLA-A2 subgroup showed increased ICT-107 activity immunologically with a tendency for improved clinical outcome |
| NCT03018288 (HSPPC-96) | Double-blind, randomized study of RT + TMZ and pembrolizumab +/− HSPPC-96 vaccine in newly diagnosed GBM | 2 | 108 | 1 year OS | Ongoing study, estimated completion 01/2025 | ||||
| Non-randomized trials | |||||||||
| NCT02149225 (GAPVAC) | GAPVAC1 and 2, GM-CSF and Poly-ICLC and TMZ in newly diagnosed GBM | 1 | 16 | AEs | 29 mo | 12 mo | Able to generate a strong and lasting immune response | Unmutated APVAC1 antigens elicited sustained responses of central memory CD8+ T cells. APVAC2 induced predominantly CD4+ T-cell responses of T helper 1 type against predicted neoepitope | |
| NCT02924038 | Varlimumab (CDX-1127) + IMA950/polyICLC in newly diagnosed GBM | 1 | 30 | Aes, CD4+, CD8+, and T-cell responses | Ongoing study, estimated completion 12/2022 | Varlimumab (CDX-1127) is an anti-CD-27 antibody that activates T cells | |||
| NCT02287428 (NeoVax) | Personalized neoantigen cancer vaccine (neoVax) + RT + pembrolizumab in newly diagnosed GBM | 2 | 56 | Aes; no. of patients with actionable peptides; no. of pts able to recieve post-RT vaccine therapy | Ongoing study, estimated completion 01/2025 | ||||
| NCT02287428 (NeoVax) | Personalized neoantigen cancer vaccine (neoVax) + RT in newly diagnosed GBM | 1b | 8 | Safety and feasibility | 16.8 mos | 7.6 mos | Neoantigen selection is feasible and induces immune response | Neoantigen-specific T cells from the peripheral blood could migrate into an intracranial glioblastoma tumour | |
| NCT02960230 | H3.3K27M peptide vaccine in children with newly diagnosed DIPG/gliomas | 1 | 29 | Aes; overall OS at 12 months | Ongoing study, estimated completion 01/2023 | ||||
| Oncolytic viral therapies | |||||||||
| Phase III trials | |||||||||
| NCT02414165 (Toca 5) | Toca 511 (retroviral replicating vector encoding cytosine deaminase + Toca FC (flucytosine) vs. lomustine, TMZ, or bevacizumab in recurrent HGG | 2/3 | 403 | OS | 11.1 mos in treatment arm vs. 12.2 in control arm (HR = 1.06, p = 0.6154) | Stopped prematurely for lack of efficacy | Data available only from company communication | ||
| Nonrandomized trials | |||||||||
| NCT01470794 | Toca 511 (retroviral replicating vector encoding cytosine deaminase + Toca FC (flucytosine) in recurrent HGG | 1 | 58 | MTD, dose-limiting toxicities | 14.4 mos | Durable complete responses were observed | |||
| NCT1491893 (PVSRIPO) | Recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO) in reccurent HGG | 1 | 61 | MTD, dose-limiting toxicities | 12.5 months (95% CI, 9.9 to 15.2) | 21% long-term survivors at 36 months | |||
| NCT02197169 (TARGET-I) | DNX-2401 ± interferon gamma (IFN-γ) for recurrent glioblastoma | 2 | 27 | No benefit with the addition of IFN/IFN poorly tolerated | Data available from ASCO poster only | ||||
| NCT00805376 (DNX-2401) | DNX-2401 (conditionally replication-competent adenovirus) +/− surgery in recurrent HGG | 1 | 37 | MTD | 9.5 mos | Long-term survivors reported | Treatment induced tumor infiltration by CD8+ and T-bet+ cells | ||
| NCT02798406 (CAPTIVE) | DNX-2401 (conditionally replication-competent adenovirus) + pembrolizumab in recurrent GBM | 2 | 49 | Objective response rate | Ongoing study, expected completion 08/2023 | ||||
| NCT02986178 | Recombinant nonpathogenic polio-rhinovirus chimera (PVSRIPO) in recurrent malignant glioma | 2 | 122 | Objective radiological response rate at 24 and 36 months | Ongoing study, expected completion 07/2021 | ||||
| NCT03896568 (Ad5-DNX-2401) | Ad5-DNX-2401 (oncolytic adenovirus) in bone marrow human mesenchymal stem cells in recurrent HGG | 1 | 36 | MTD | Ongoing study, estimated completion 05/2022 | ||||
| NCT01956734 (DNX2401) | DNX-2401 + temozolomide in recurrenct glioblastoma | 1 | 31 | Nb of participants with AEs | Study completed 2018, no info available | ||||
| NCT02026271 | Ad-RTS-hIL-12 with veledimex in recurrent HGG | 1 | 38 | Safety and tolerability of varying doses of intratumoral Ad-RTS-hIL-12 and oral veledimex | 12.7 mos | Response correlated with CD8+ (cytotoxic) and FoxP3+ (regulatory) T-cell counts in the peripheral blood | |||
| NCT03330197 | Ad-RTS-hIL-12 + veledimex in pediatric subjects With brain tumors including DIPG | 1 | 25 | Safety and tolerability | Study ongoing, expected completion 12/2022 | ||||
| NCT00390299 | Carcinoembryonic antigen-expressing measles virus (MV-CEA) in trecurrent glioblastoma multiforme | 1 | 23 | DLT | |||||
| NCT03294486 | Safety and efficacy of the oncolytic virus armed for local chemotherapy, TG6002/5-FC, in recurrent GBM | 1 | 78 | DLT | |||||
| NCT02457845 (G207) | HSV G207 (oncolytic HSV-1) + RT; children with recurrent HGG | 1 | 18 | MTD | Enrollment completed 1/2021, results not yet availabe | ||||
| NCT03152318 (rQNestin) | rQNestin34.5v0.2 (oncolytic HSV-1) + cyclophosphamide in recuurent HGG | 1 | 108 | MTD | Ongoing study, estimated completion 07/2022 | Ongoing study | |||
| NCT00390299 | MV-CEA (carcinoembryonic antigen expressing measles virus) in recurrent GBM | 1 | 23 | MTD, severity of Aes, overall toxicity | Accrual completed | ||||
| NCT01301430 (ParvOryx01) | H-1 PV in recurrent HGG | 1 | 18 | Safety and tolerability | |||||
| NCT03714334 | DNX-2440 conditionally replication-competent adenovirus with OX40 ligand (T-cell stimulator) in recurrent GBM | 1 | 24 | Treatment-related Aes | |||||
| NCT02062827 (M032-HSV-1) | M032-HSV-1 (second-generation oncolytic HSV with IL-12 (immune stimulatory) in recurrent GBM | 1 | 36 | MTD | |||||
| Checkpoint inhibitors | |||||||||
| Phase III trials | |||||||||
| NCT02017717 (Checkmate 143) | Nivolumab vs. bevacizumab in recurrent GBM | 3 | 626 | OS | OS: 9.5 mo vs. 9.8 mo (NS) | ||||
| NCT02617589 (Checkmate 498) | Nivolumab + RT vs. RT + TMZ in MGMT unmethylated newly diagnosed GBM | 3 | 550 | OS | |||||
| NCT02667587 (Checkmate 548) | Nivolumab + RT-TMZ vs. RT + TMZ in MGMT methylated newly diagnosed GBM | 3 | 693 | OS | |||||
| Nonrandomized trials | |||||||||
| NCT02336165 | Durvalumab (MEDI4736) in newly diagnosed and recurrent glioblastoma (5 non comparative arms) | 2 | 159 | OS at 12 mos | Ongoing study | ||||
| NCT02054806 | Pembrolizumab (MK-3475) in advanced solid tumors | 1b | 26 GBMs | Best overall response | 14.4 mos | 2.8 mos | |||
| NCT02054806 | Pembrolizumab in recurrent GBM | 2 | 26 | 4% partial responses/48% SD | |||||
| NCT02337491 | Pembrolizumab alone; pembrolizumab + bevacizumab in recurrent GBM | 2 | 80 | Pembrolizumab maximum tolerated dose; pembrolizumab dose-limiting toxicity at 6 mos/PFS | Accrual completed | ||||
| NCT2313272 | Hypofractionated stereotactic RT + pembrolizumab + bevacizumab in recurrent HGG | 1 | 32 | MTD | |||||
| CAR T-Cell therapies | |||||||||
| Nonrandomized trials | |||||||||
| NCT2208362 | IL13Ralpha2specific CAR T cells in recurrent HGG | 1 | 92 | Aes (grade ≥3) | Ongoing study | ||||
| NCT02209376 | EGFRvIII CAR T cells in EGFRvIII positive GBMs | 1 | 11 | Aes at 2 years | Study prematurely terminated | ||||
| NCT01109095 | HER2 virus-specific CAR T cells | 1 | 16 | DLTs | Ongoing study | ||||
| NCT02442297 | HER2 CAR T cells | 1 | 28 | DLTs | Ongoing study | ||||
| NCT00331526 | Cellular adoptive immunotherapy in recurrent GBM | 2 | 33 | Aes/PFS & OS | 12.05 mos | ||||
| Combined approaches | |||||||||
| Randomized trials | |||||||||
| NCT02866747 (STERIMGLI) | Hypofractionated stereotactic radiation therapy ± durvalumab in recurrent GBM (STERIMGLI) | 1/2 | 112 | DLT (phase 1)/OS (phase 2) | ongoing study, completion expected 12/2024 | ||||
| Nonrandomized trials | |||||||||
| NCT02960230 | H3.3K27M peptide vaccine + nivolumab in children with newly diagnosed DIPG/gliomas | 1/2 | 49 | Safety of the vaccine in combination with nivolumab | Ongoing study, estimated completion 01/2023 | ||||
| NCT02648633 | Stereotactic radiosurgery with nivolumab and valproate in patients with recurrent glioblastoma | 1 | 4 | Feasability | |||||
| NCT02311582 | Pembrolizumab + MRI-guided laser ablation in recurrent malignant gliomas | 1/2 | 58 | MTD (phase 1)/PFS (phase 2) | Ongoing study, expected completion 12/2024 | MLA aims at disrupting the blood–brain barrier | |||
| NCT01811992 | Combined cytotoxic and immune-stimulatory therapy for glioma | 1 | 19 | MTD | Ongoing study, expected completion 04/2021 | ||||
| NCT01205334 (COGLI) | CMV-specific cytotoxic T cells in recurrent GBM | ||||||||
| NCT02798406 (CAPTIVE) | Combination adenovirus + pembrolizumab to trigger immune virus effects in recurrent GBM (CAPTIVE) | 2 | 49 | Objective response rate | Ongoing study, enrollment completed 03/21 | ||||
| Modification of the tumor microenvironment | |||||||||
| Nonrandomized trials | |||||||||
| NCT02052648 | IDO inhibitor + temozolomide in recurrent HGG | 1/2 | 160 | Dose determination and 6-month PFS | Accrual completed | Indoximod is an immunometabolic adjuvant that induces T-cell activity in cancer | |||
| NCT02526017 | Cabiralizumab in combination with nivolumab in patients with selected advanced cancers (FPA008-003) | 1 | 295 | Safety | Accrual completed | Cabiralizumab is a humanized monoclonal antibody directed against the tyrosine kinase receptor colony stimulating factor 1 receptor (CSF1R; CSF-1R) | |||
5.2. Oncolytic Viruses
5.3. Checkpoint Inhibitors
5.4. Adoptive Cell Transfer and CAR T Cells
5.5. Identified Limits and How to Overcome Them
This entry is adapted from the peer-reviewed paper 10.3390/cancers13153721