The paired-related homeobox gene (PITX2) has been considered to be a potential gene that may trigger AF risk variants on chromosome 4q25and two SNPs (rs2200733 and rs10033464) in chromosome 4q25 were reported. Although various advanced technologies have been developed, their effectiveness is limited and existing treatment regimens are rarely curative.
- cardiac arrhythmias
- atrial fibrillation
- PITX2
- computational model
- electrical remodelling
- structural remodelling
- calcium handling
- mRNA
- electrophysiology
1. Introduction
Atrial fibrillation (AF), characterized by rapid and disorganized electrical activation in the upper chambers of the heart, is the most common cardiac arrhythmia [1]. AF affects 2–3% of the population and its prevalence is rising with age from 2.4% in people 65 years old to 10% in those older than 80 years [2]. Recently, a large number of studies have shown that AF, especially lone AF, has an important genetic component [3].
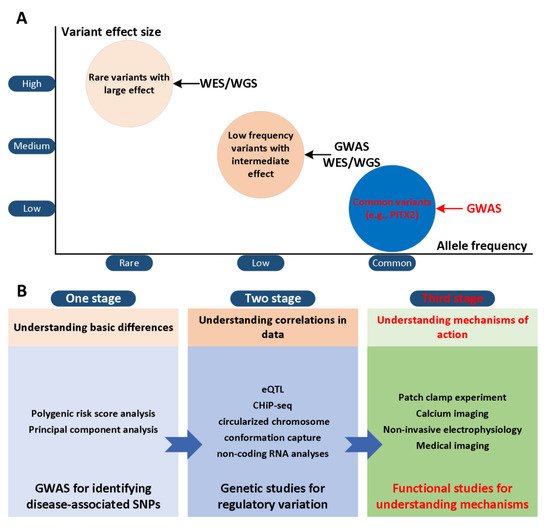
The paired-related homeobox gene (PITX2) has been considered to be a potential gene that may trigger AF risk variants on chromosome 4q25 [4,5] and two SNPs (rs2200733 and rs10033464) in chromosome 4q25 were reported [6]. Although various advanced technologies have been developed, their effectiveness is limited and existing treatment regimens are rarely curative [7,8,9]. This result is partly due to our limited understanding of the mechanisms of AF in the first and second stages ( Figure 1 A). In the third stage, the development of actionable personalized approaches, which take into account patient-specific profiles and arrhythmia mechanisms, will likely be essential to overcome current challenges in AF management ( Figure 1 B). Computational modelling and simulation are indispensable in cardiac electrophysiology in the study of complex arrhythmias [10,11,12], such as AF. The multi-scale model of cardiac electrophysiology provides a framework that can integrate experimental and clinical findings [13], and link micro-scale phenomena to emerging behaviors of the entire organ [14]. Computational modeling is now an important part of AF mechanism research, because it can supplement experimental observations and suggest novel mechanisms [15,16,17,18,19,20,21,22,23,24,25,26]. In addition, the entire atrial simulation is currently used to design novel and personalized treatment strategies, thereby contributing to the development of precision medicine in cardiology. In this narrative review, we focus on the latest developments in the atrial model in elucidating the mechanism of PITX2-dependent AF. We summarize experimental studies of the role of PITX2 in cardiogenesis and arrhythmogenesis, advances in atrial modelling, and modelling studies for investigating PITX2-dependent AF mechanisms. Specifically, we summarize the progress in the development of multi-scale AF models, and then focus on the mechanistic connection between alternations in atrial structure and electrophysiology with PITX2-dependent AF from the perspective of computational modeling. Researchers focus on how AF modeling can supplement experimental data in ways that cannot be achieved outside of the simulation framework, and how AF models can reveal novel AF mechanisms. Researchers conclude the review with a summary of the future prospects of the atrial model’s mechanical understanding of AF, towards the goal of understanding patient-specific AF mechanisms that would allow for personalised treatment.
2. The Role of PITX2 in Cardiogenesis and Arrhythmogenesis
In the early stage of embryogenesis, PITX2 is directly regulated by the nodal-mediated left-right asymmetry pathway, thereby imparting left morphogenesis to several organs in the body [36]. During heart development, PITX2 is essential for left-right asymmetry, differential regulation of left ventricular identity, and ventricular asymmetric remodeling procedures [34]. PITX2 also confers left atrial morphology [37]. The left atrium with a null mutation of PITX2 has morphological features on the right side, including venous valves and trabecular myocardium [37]. In addition, PITX2-deficient embryos also have bilateral or ectopic sinus nodes, which may explain the AF susceptibility observed in adult animals with decreased PITX2 expression [38,39,40]. PITX2 also plays a vital role in regulating the development of the cardiac conduction system and pulmonary myocardium [35,39].
Several potassium channel, sodium channel and calcium handling genes are regulated by PITX2, as seen in mutant PITX2 models. TASK-like background currents, contributors to the resting membrane potential, decreased in PITX2c +/− mice [56]. In the left atrial chambers, Kir2.1 (Kcnj2) and Nav1.5 (Scn5a ) channel expression are decreased [57], while Bmp10 is highly upregulated [58]. These findings support that PITX2 may control atrial chamber dimensions, because overexpression of Bmp10 plays a crucial role in regulating physiological hypertrophy. In the right atrial myocytes, overexpression of PITX2c increased I Ks density and reduced I CaL density [59]. In additional to changes in ion channels, impaired Gja5 expression was found in adult heterozygous PITX2 mutant mice [42,57,60]. Moreover, several calcium handling genes were found to be regulated by PITX2. In atrial-specific NppaCrePitx2 mice, Cacna1c is significantly down-regulated, whereas Atp2a2, Casq2, and Pln display a major and RyR2 a minor up-regulation [44]. Recently, similar defective calcium homeostasis links the 4q25/PITX2 variants to the risk of AF [41,61]. Experimental results showed that human atrial myocytes from patients carrying the PITX2 variant (rs13143308T) display increased Serca2a expression, SR calcium load, and RyR2 phosphorylation, which likely may cause triggered activity observed in these patients [61].
It has been found that both overexpression and inhibition of PITX2 are related to human AF. In patients who need ablation of AF, PITX2 is expressed in a wide range in the entire tissue sample of the left atrial appendage and the gradient of PITX2 mRNA is increased [58]. PITX2 concentrations in whole left atrial tissue were similar in patients with and without AF recurrence. Morphological analysis of the patient’s left atrial appendage tissue biopsy showed that the tissue heterogeneity of some samples had obvious fatty deposits and fibrosis, while other samples had high myocardial content. However, the concentration of PITX2 in left atrial appendage cardiomyocytes of patients with recurrent AF is lower than that of patients without relapse. Therefore, the recurrence of AF is related to the decrease of PITX2 concentration in myocardial cells but not the entire left atrial appendage tissue PITX2. This study supports the hypothesis that decreased PITX2 concentration in left atrial cardiomyocytes is associated with recurrence of AF. Furthermore, patients with chronic AF have a higher concentration of PITX2 in the myocardial cells of the right atrial appendage compared with sinus rhythm [59]. Further analyses showed that the expression of PITX2 was positively and negatively correlated with I Ks and I CaL densities, respectively. These results supported that impaired PITX2 in right atrial appendage cardiomyocyte was associated with electrical remodelling, contributing to AF.
In addition to calcium handling and electrical remodelling, PITX2-dependent structural remodelling includes alteration in gap junction and its distribution, fibrosis and cell proliferation [66]. The decrease of PITX2 concentration in myocardial cells is related to the recurrence of AF. Compared with patients with no recurrence of AF, patients with recurrent AF have a lower concentration of PITX2 in left atrial appendage myocardial cells. In the patient’s left atrial appendage tissue , obvious fat deposition and fibrosis also were found in some specimens, while high myocardium content was observed in other specimens. Up-regulated Bmp10 linked structural remodelling to imparied PITX2 [58], because the overexpression of Bmp10 plays a key role in regulating physiological hypertrophy [67]. Experimental results also showed that the downregulation of Pitx2 was inversely correlated with the upregulation of miR-21 which may play a vital role in the development of fibrosis by promoting the proliferation of interstitial fibroblasts and increasing the abnormal deposition of the extracellular matrix [68]. Therefore, reduced PITX2 may contribute to the development of fibrosis. PITX2 also regulated the cell-cell gap junction gene (Gja1, Gja5). The expression of Gja1 (Cx43) was highly up-regulated, while the expression of Gja5 (Cx40) was severely down-regulated in the left atrial chamber of the atrial-specific Pitx2 mutant. In addition, Gja1 (but not Gja5) showed a dose-dependent regulation of PITX2 and was upregulated in both left and right atrial chambers [44]. Clinical studies support this notion changes in gap junctions arising from impaired PITX2 may influence electrical conduction. Carriers had relatively increased conduction velocity heterogeneity, complex signals, regional left atrial slowing, or conduction block particularly in the posterior and lateral walls [64].
3. Mechanistic Insights into PITX2-Dependent AF Using Computational Models
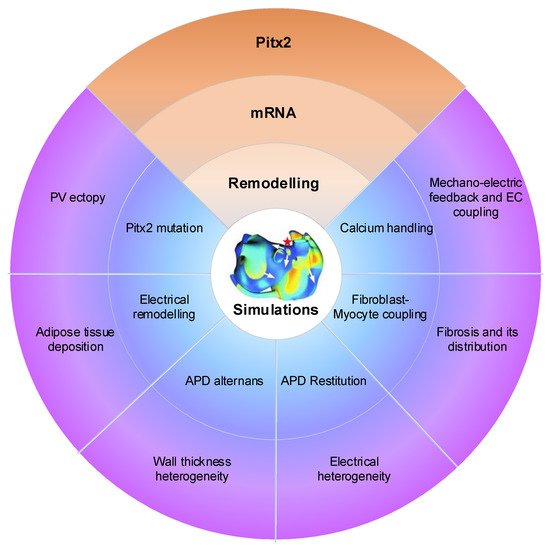
Atrial computational modelling allows integration of detailed basic information about atrial electrophysiology at the cellular and tissue level and relevant simulations provides insight in the fundamental mechanisms involved in initiation and perpetuation of PITX2-dependent AF. Increased triggered activity is the primary mechanism of PITX2-dependent AF initiation. Maintenance of PITX2-dependent AF is associated with remodelling (including electrical and structural remodelling) of the atria. Regions of structural or functional inhomogeneities arising from left–right asymmetry of Pitx2 expression may promote maintenance of re-entrant drivers. Experimental data on PITX2-dependent remodelling have been incorporated in atrial models to predict arrhythmic behaviour, linking the impaired PITX2 to AF with the altered electroanatomical substrate. Key structural and functional alternations that are mechanistically linked to PITX2-dependent AF and have been studied using computational models are calcium handling, PITX2 mutation, electrical remodelling, electrical heterogeneity, APD restitution and atrial wall thickness heterogeneity ( Figure 10 ). Although pulmonary vein (PV) ectopy [119], atrial adipose tissue infiltration, development of repolarisation alternans, presence of atrial fibrosis and its distribution, fibroblast-myocyte uncoupling, and atrial stretch with mechano-electrical feedback have a significant role in the maintenance of re-entrant drivers, current atrial modelling studies have not incorporated these alterations.
Atrial modelling studies on AF have demonstrated that action potential shortening contributes to PITX2-induced AF [19,21,22,91]. Consistent with experimental data on PITX2 insufficiency-induced electrical remodelling, remodelled targets include I K1 , I Na , I CaL , I Ks and gap junctions. In these remodelled targets, we observed that downregulated I K1 resulted in a more positive resting membrane potential and a prolonged action potential duration, upregulated I Na contributed to a high overshoot and the maximum depolarization rate, the I Ks increase and (or) the I CaL decrease led to a reduction in action potential duration and reduced gap junctions caused slow conduction [21]. The genesis of PITX2-dependent AF may be attributable to action potential duration shortening due to changes in I Ks and I CaL and slow conduction resulted from downregulated gap junctions. The combined impact of electrical remodeling on initiation and maintenance of atrial fibrillation can be characterized by atrial tissue’s vulnerabilities to re-entry. On the one hand, PITX2-dependent electrical remodelling increased the spatial vulnerability of atrial tissue by decreasing the substrate size and wavelength due to abbreviated the effective refractory period and reduced conduction velocity. On the other hand, the temporal vulnerability of atrial tissue is measured as the vulnerable window during which unidirectional conduction block and re-entry can be induced by a test stimulus. And the vulnerable window was significantly increased for atrial tissue incorporating remodeling. Consequently, the increase of temporal and spatial vulnerabilities of atrial tissues may explain why the PITX2-dependent electrical remodelling promotes initiation of re-entry.
In addition to AF initiation, the PITX2-dependent electrical remodelling promoted the maintenance of reentry by flattening the action potential duration restitution curves [22]. In the case of the PITX2-dependent electrical remodelling, the maximum slope of the action potential duration restitution curve was reduced. This led to a gradual decrease in action potential duration alternans toward a steady state level and thereby a stable spiral wave at the tissue level. The stable spiral wave was characterized by decreased tip meander area and prolonged life span.
Increased electrical heterogeneity between right atrium (RA) and left atrium (LA) due to down-regulated PITX2 expression implicated in the initiation and maintenance of re-entrant arrhythmias [21]. Since the ratio of PITX2 between LA and RA is 100:1 in the human atria, the extent of remodelling after PITX2 deletion is dependent on the amount of PITX2. Increased electrical heterogeneity may result from the difference in PITX2-induced remodelling between LA and RA. In our simulation study, we observed that electrical remodeling induced by PITX2 deficiency causes action potential duration abbreviation and ectopic depolarizations in LA myocytes, but not in RA myocytes. Thus, PITX2 insufficiency can cause an elevated difference in electrical properties between RA and LA cells, increasing repolarization dispersion in tissue and thereby susceptibility to the development of re-entry. Furthermore, Pitx2 deficiency can also cause LA structural remodeling by regulating cardiac structural genes, increasing electrical and structural heterogeneity between the two atrial chambers. The presence of increased fibrosis and decreased cell-to-cell coupling under structural remodelling further facilitates slow conduction, wavelength abbreviation, triggered activity and the initiation of re-entrant drivers, increasing susceptibility to AF. It is known that PITX2-dependent network regulates cardiac structural genes (Gja1, Gja5 and Dsp) and PITX2-induced structural remodeling leads to fibrosis and cell-cell uncoupling. Also, structural remodeling due to the left-sided PITX2 expression increases the intrinsic heterogeneity (i.e., conduction velocity and wavelength) between RA and LA, facilitating the development of re-entry.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22147681