Extracellular vesicles (EVs) are membrane-bound bilayered lipid particles that are secreted from both prokaryotic and eukaryotic cells, carrying a cargo of biological molecules (i.e., protein, nucleic acid, lipids and metabolites) from their parent cells. Dental tissue-derived (or stem) cells have remarkable characteristics for therapeutic application, being easily accessible and a rich source of stem cells with a well-known regenerative capacity. A great variety of multipotent adult or postnatal stem cells can be retrieved from dental tissues, especially from periodontal tissue and dental pulp from extracted permanent teeth (dental pulp stem cells—DPSCs) and exfoliated deciduous teeth (SHED). Stem cells can be obtained from dental apical papilla tissues (SCAP) and dental follicles (DFSCs, or DFCs) of the developing tooth. Importantly, EV that is derived from these cells can be detected within periodontal tissues and biofluid (i.e., gingival crevicular fluid).
1. Introduction
Extracellular vesicles (EVs) are membrane-bound bilayered lipid particles that are secreted from both prokaryotic and eukaryotic cells, carrying a cargo of biological molecules (i.e., protein, nucleic acid, lipids and metabolites) from their parent cells [
1]. Initially, EVs were considered ‘cellular dust’, generated by cellular metabolism, until their biological role in the mineralization of bone was recognized [
2,
3]. A principal role of EVs is as an intercellular communicator of biological information into a recipient cell. This interaction can trigger signaling cascades and modulate cell behavior [
4]. The biological function of EVs is defined by the parent cells from which they originate. EVs are involved in almost all cellular interactions, especially tumor metastasis, tissue homeostasis, and inflammatory regulation [
4,
5]. Due to their constituent biological molecules, EVs hold great promise as a therapeutic delivery system in regenerative medicine.
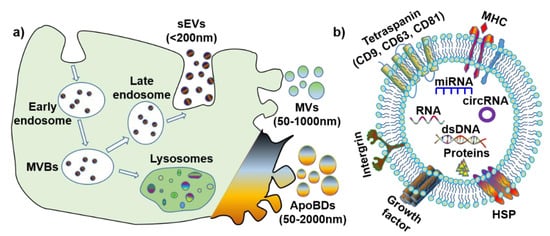
The definition, terminology and subtypes of EVs are still being debated. The International Society of Extracellular Vesicles (ISEV) recommends a division of EV subtypes based on their size: medium/large EV (>150 nm) and small EV (<150 nm) [
6]. However, considering the discrepancies in the published literature, for simplification purposes, this review will define EV subtypes based on both their size and biogenesis (
Figure 1a): small extracellular vesicles (also known as exosomes) (sEVs, <200 nm), microvesicles (MVs, 50–1000 nm) and apoptotic bodies (ApoBDs, 50–2000 nm). Furthermore, it is noteworthy that all EVs have various membrane proteins (e.g., tetraspanin, MHC, and HSP) and components (e.g., dsDNA, RNA, microRNA, circular RNA [
7], and proteins) (
Figure 1b).
Figure 1. The biogenesis and contents of extracellular vesicles (EVs). (a) Biogenesis and size of three EV subtypes. (b) Common surface markers and cargos of EVs. sEVs: small extracellular vesicles; MVs: microvesicles; ApoBDs: apoptotic bodies; MVBs: multi-vesicular bodies; MHC: major histocompatibility complex; HSP: heat-shock protein; dsDNA: double-stranded DNA; miRNA: microRNA; circRNA: circular RNA.
1.1. Small EV (Exosomes)
Small EVs (sEV), or exosomes, originate from endosomes, and are biological nanoparticles that are smaller than 200 nm [
5]. Further, sEVs are produced through an endocytic pathway, and their particle size is partially overlaid with that of microvesicles and apoptotic bodies. The biogenesis process for sEV is unique, whereby the endosomal network is the source of sEV that produce, classify, distribute and define the proper destination of the secreted sEV [
2,
8]. Endosome production can be categorized into the following three subtypes, according to each stage of development: early endosomes, late endosomes, and recycling endosomes. Early endosomes are formed by inward budding of the cell membrane, before a second inward budding of the endosomal membrane that results in the formation of late endosomes—intraluminal vesicles (ILVs). Late endosomes containing IVLs are named multi-vesicular bodies (MVBs), and the MVBs either fuse with lysosomes to degrade or follow the endocytic pathway for sEV generation. Once fusion with the plasma membrane is completed, the small membrane-enclosed vesicles are released into the extracellular matrix.
The biogenesis of sEV is affected by the following two main pathways that can induce multi-vesicular bodies (MVBs) generation: the endosomal sorting complex, required for the transport (ESCRT)-dependent pathway and ESCRT-independent pathway [
9]. For the ESCRT-dependent pathway, ESCRT I and ESCRT II mediate the invagination of the late endosomal membrane, and ESCRT III will be recruited to the invaginated membrane sites. The cargo proteins are then deubiquitinated, and this stimulates the departure of the vesicle and the formation of MVBs. In the ESCRT-independent pathway, the neutral sphigomylinase2 (nSMase2) takes sphingolipids as substrates and converts sphingolipids to ceramide at the endosomal membrane. Following this, the microdomain is prepared for merging into a larger structure, which accelerates the endosomal budding and biogenesis of MVBs [
10]. Moreover, sEVs that are produced by these different pathways possess different biomarkers, except CD63, which is the most common biomarker for all sEVs [
11,
12]. With respect to the ESCRT-dependent pathway, if endocytosis is mediated by Ras-related protein 27A/B (RAB27A/B), TSG101 is a biomarker of sEV. If the endocytosis is mediated by phospholipase D2 (PLD2) and RAB7, through the ESCRT-independent pathway, the biomarkers of sEV are alix, syntenin, and syndecan. As for the RAB11/35-mediated ESCRT-independent pathway, CD81, Wnt and proteolipid protein (PLP) are the preferred biomarkers.
The function of sEV in intercellular communication is determined by the interconnection between sEV surface proteins and receptors on the recipient cells that subsequently activates a variety of signaling pathways [
5]. Further, sEVs arising from different cell types have different cargos that dictate and direct different biological effects. The sEVs are highly abundant in biofluids [
13,
14,
15], and they have been demonstrated to be associated with immune response, viral pathogenicity, osteogenesis, odontogenesis, neuroprotection, angiogenesis, and anti-tumor functions [
16]. For example, oral cancer cell-derived sEVs create a mechanism that can promote tumor progression by modifying vesicular contents and establishing a distant premetastatic niche with molecules that favor cancer cell proliferation, migration, invasion, metastasis, angiogenesis, and even drug resistance [
17]. Evidence that sEVs play an important role in cell differentiation suggests that sEVs may have a potential role in tissue regeneration.
1.2. Microvesicles
Microvesicles (MVs) are membrane vesicles of different sizes, surrounded by a lipid layer of membrane, and they range in size from 50 nm to 1 μm. Microvesicles are generated by the outward budding of the plasma membrane, and are abundant in tissues/cells and biofluids [
18]. The contents of MVs are similar to that of sEV. The MV components of note include CD40, selectins, integrins, cytoskeletal proteins, and cholesterol [
19].
The biogenesis of MV involves the contraction of cytoskeletal proteins and phospholipid redistribution, contributing to a dynamic interplay in the plasma membrane and the resultant formation of microvesicles. Within the plasma membrane, the aminophospholipid translocase regulates phospholipid distribution, transferring phospholipids from one leaflet to another. Once phosphatidylserine (PS) is translocated to the leaflet of the outer membrane, the outward blebbing of the membrane and microvesicle formation is initiated. The interaction between actin and myosin causes the cytoskeletal structure contraction, which mediates membrane budding [
20].
MVs have been reported to maintain tissue homeostasis during tissue regeneration, angiogenesis, anti-tumor effects, and in pathologies such as tumorigenesis, chronic inflammation, and atherosclerosis [
19]. MVs that are produced by blood cells (e.g., neutrophils, macrophages, and platelets) are involved in the pro-coagulatory response [
21]. MVs can be both pro-inflammatory and anti-inflammatory; this is determined by the induction or stimulation that is received by their parent cells. MVs that are produced by tumor cells enhance invasiveness and accelerate cancer progression, as well as strengthen the drug resistance of tumor cells [
22]. This indicates that MVs are potential therapeutic agents for tissue regeneration; however, the function of MVs in periodontal tissue healing and regeneration requires further investigation.
1.3. Apoptotic Bodies
Apoptotic bodies (ApoBDs) are produced by cells undergoing apoptosis, and vary in size from 50 nm to 2 µm [
23,
24]. ApoBDs result from the formation of subcellular fragments when an apoptotic cell disassembles. They are comprised of molecular components from living cells and provide a rich molecular pool for recipient cells. However, ApoBDs are engulfed by macrophages and digested by phagolysosomes shortly after they are released [
25]. ApoBDs and apoptosis are not related to an inflammatory reaction, the constituents in dying cells and ApoBDs are not released automatically to the environment, and anti-inflammatory cytokines are not generated during engulfing. ApoBDs have phosphatidylserine (PS) on their surfaces, to attract engulfing cells, and are considered to be specific biomarkers for ApoBDs [
26]. Autoimmune diseases may be associated with defects in the clearance of ApoBDs. ApoBDs may stimulate the formation of thrombus and improve anti-cancer immunity.
Increasing evidence suggests that ApoBDs have important immune regulatory roles, in autoimmunity, cancer, and infection [
24], as well as promoting osteogenesis [
27]. For example, ApoBDs that are derived from mature osteoclasts can induce osteoblast differentiation by activating the protein kinase B/phosphoinositide 3-kinases (PI3K/AKT) pathway [
27]. However, knowledge of their function and role is still limited and more studies are required in this field.
2. The Source and Characteristics of Periodontal (Dental Pulp) Cells
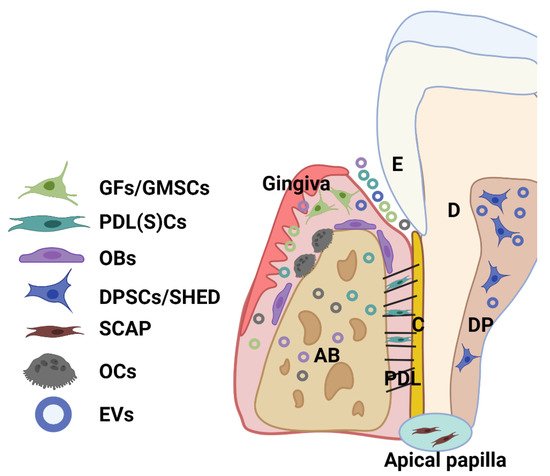
Dental tissue-derived (or stem) cells have remarkable characteristics for therapeutic application, being easily accessible and a rich source of stem cells with a well-known regenerative capacity. A great variety of multipotent adult or postnatal stem cells can be retrieved from dental tissues, especially from periodontal tissue and dental pulp from extracted permanent teeth (dental pulp stem cells—DPSCs) and exfoliated deciduous teeth (SHED). A healthy periodontium consists of soft (periodontal ligament-PDL and gingiva) and hard (alveolar bone and cementum) tissue, and cells residing within the healthy periodontal tissues include periodontal ligament (stem) cells (PDLSCs), PDL and gingival fibroblasts (PDLF, GFs), or gingival stem cells (GMSCs), osteoblasts (OBs), osteoclasts (OCs), and various immune cells (
Figure 2) [
28,
29]. Moreover, stem cells can be obtained from dental apical papilla tissues (SCAP) and dental follicles (DFSCs, or DFCs) of the developing tooth [
28,
29]. Importantly, EV that is derived from these cells can be detected within periodontal tissues and biofluid (i.e., gingival crevicular fluid) (
Figure 2).
Figure 2. Schematic showing the main cell population and cell products (EVs) within a healthy periodontium. Various cells reside in the periodontium, such as periodontal ligament (stem) cells (PDLSCs), fibroblasts (GFs) and stem cells (GMSCs) from the gingiva, osteoblasts (OBs), osteoclasts (OCs), and various immune cells. AB: alveolar bone; C: cementum; D: dentin; DP: dental pulp; PDL: periodontal ligament; SHED: dental pulp cells from human exfoliated deciduous teeth (SHED); SCAP: cells from periodontal apical papilla tissues (SCAP).
Dental mesenchymal stem cells originate from the neural crest ectomesenchyme and reside in stromal niches (perivasculature and peripheral nerve-associated glia cells). The current consensus holds that both perivascular cells [
30] and glia cells [
31] are responsible for dental MSCs origin, as revealed in mouse experiments [
31]. Much like bone-marrow-derived MSCs that originate from mesoderm [
32], dental stem cells express MSCs markers and exhibit multipotent linage regeneration (i.e., osteogenic, chondrogenic, neurogenic) and immunomodulatory capabilities. These properties make these cells suitable candidates for therapeutic application (reviewed by Chalisserry et al. in [
33]) in neurological disorders, angiogenesis, dentin-pulp regeneration and periodontal regeneration. PDLSCs, GFs, DPSCs, SHED, and DFSCs have been demonstrated to promote multiple-tissue regeneration, both
in vitro and
in vivo [
34,
35,
36,
37,
38,
39,
40]. However, cell therapy has several challenges, including high cost, insufficient cell number, and associated regulatory barriers. On the other hand, a cell-free approach, centered around cell products (i.e., EVs derived from these cells), has been proposed, and there is an emerging focus on cell-derived EVs as potential therapeutic agents to promote periodontal regeneration. The utilization of sEVs for dental tissue regeneration is emerging as a viable cell-free treatment option, with ‘proof of concept’ studies reported using bone marrow or adipose MSC-derived sEVs (reviewed in [
41,
42,
43]); yet, periodontal or dental pulp cell sources are likely to uniquely reflect the functional complexity of the periodontium and oral cavity.
3. Cell-Derived sEV Isolation Methods
3.1. General Concepts
Although sEVs have been studied for decades, there is still no standardized protocol for their isolation. Despite the presence of recommended guidelines for EV isolation and characterization, such as the Minimal Information for Studies of Extracellular Vesicles 2014 (MISEV2014) and MISEV 2018, these guidelines are not always followed.
Prior to the isolation of sEV, sequential centrifugation is commonly used to remove cell debris and large EVs, as follows:
-
Step 1: the cell conditional media (CM) is harvested and centrifuged at 300–400× g to remove cells, and the supernatant (SN) is collected;
-
Step 2: the SN collected in step 1 is centrifuged at 2000–3000× g to remove cells debris and apoptotic bodies. The SN is collected from this step;
-
Step 3: SN from step 2 is centrifuged at 10,000–20,000× g to remove the aggregates of biopolymers, microvesicles, and the other structures with a buoyant density higher than sEVs. The SN is collected from this step;
-
Step 4: then, the following isolation methods are used to enrich the sEVs: ultracentrifugation, sucrose gradient centrifugation, size exclusion chromatography, precipitation-based isolation, immunoaffinity chromatography, and ultrafiltration.
Given the growing interest in EVs, technical standardization is critical, as many different methodologies have been utilized for isolation and analysis. The influence of these various techniques on the downstream composition and functionality of EV cargos remains unclear; accordingly, the ISEV position papers [
6,
44] have raised the need to define ‘good practices’ and ultimately archive standardization. However, many researchers are not following these four steps, due to a lack of standardized protocols. Here, our review briefly introduces each isolation method, and discusses its merits and disadvantages (listed in
Table 1).
Table 1. Representative advantages and disadvantages of various EV isolation methods.
| Method |
Time |
Advantages |
Disadvantage |
Ultracentrifuge
(100,000×–200,000× g for 1–2 h |
1.5 h to 10 h |
|
|
| Floatation-related methods (sucrose gradient centrifugation) |
250 min to 1 day |
-
Pure EV population
-
No protein contamination
|
• Fails to separate large vesicles with similar sedimentation rates |
| Size exclusion chromatography (SEC) |
~30 min (including column washing) |
-
Time-efficient
-
Pure EV product
|
• sEV and microvesicles cannot be separated |
| Precipitation based isolation (sodium acetate, PEG, protamine) |
Overnight incubation |
|
|
| Immunoaffinity chromatography |
~240 min |
• Very pure EV subpopulation (i.e., CD9+ EV) |
-
Low EV yield
-
Low scalability
|
| Membrane filtration/Ultrafiltration |
~130 min |
|
• High contamination of non-EV protein |
3.2. Ultracentrifuge
Ultracentrifugation is the gold-standard method for isolating sEV, as the equipment is relatively easy to access and the methodology is technically straightforward. The method involves an ultracentrifugation step at 100,000×–200,000×
g to pellet sEV [
45]. However, ultracentrifugation has disadvantages, in that it leads to a low recovery rate of sEV, it is time consuming (1.5–10 h), contains non-vesicular macromolecule contamination, and results in EV aggregation.
3.3. Floatation-Related Methods (Sucrose Gradient Centrifugation)
Floatation-related methods distribute molecules based on the buoyant density, and the protein aggregates and sEV can be sufficiently separated. Differential gradient centrifugation (usually takes 250 min—1 day) takes advantage of buoyant density to fractionate EVs using sucrose or idoxanol gradients [
45]. The sEVs can be separated by the discontinuous gradient sucrose solution, with each layer containing the desired size of EV. Other chemical reagents (i.e., iodixanol) can also be utilized instead of sucrose, for continuous EV harvest with no layers. Non-vesicular protein contaminants are distributed at a reduced level within this method, resulting in less protein contamination. However, sucrose gradient centrifugation cannot separate large particles that have a similar sedimentation rate.
3.4. Size Exclusion Chromatography (SEC)
SEC can be used to isolate small sEV, based on the size of the molecules, where large particles pass through the gel earlier than the small-sized molecules. The small-sized particles are trapped in the tiny pores on the surface of the gel, while the larger molecules can bypass the gel or receive less interference from the gel [
46]. This technique has been well established with commercialized SEC columns, including qEV (iZON Science), Exo-spin™ SEC columns (Cell Guidance Systems Ltd.) and Pure-EVs SEC columns (HandaBioMed, Lonza). SEC has been proposed as an effective alternative method for pure sEV isolation, with a key advantage being its time efficiency (~30 min, including 10 mL of column washing with PBS). However, the similarly sized sEV and microvesicles cannot be separated by SEC.
3.5. Precipitation-Based Isolation (Sodium Acetate, PEG, Protamine)
Precipitation-based isolation has the following two mechanisms: polymeric precipitation and neutralizing charges [
47]. In polymeric precipitation, a soluble polymer, usually polyethylene glycol (PEG), is mixed with EV samples and the mixture is incubated overnight, and EVs are sedimented by low-speed centrifugation at 1500 g. PEG precipitation enables a simple process for a large number of samples. Commercial kits, such as ExoQuick (System Biosciences), total exosome isolation reagent (Invitrogen), EXO-Prep (HansaBioMed), exosome purification kit (Norgen Biotek), and miRCURY exosome isolation kit (Exiqon), are based on this principle. For the other precipitation method, all EVs possess negative charges, so positively charged molecules (i.e., sodium acetate and protamine) are chosen for the precipitation. This method is popular due to its straightforward protocol; however, these precipitation methods lead to low sEV purity due to co-precipitation of the components from CM or biofluids, such as protein, DNA and RNA, and hence further purification is required.
3.6. Immunoaffinity Chromatography
The monoclonal antibodies (mAbs) against specific sEV surface proteins (i.e., CD 9) are fixed on the column, to capture a specific sEV population [
48]. Once the CM passes through the column, the EVs, which express certain exosomal markers on their membrane, will be captured by the mAbs. This method leads to a very pure EV population, but low yield and scalability. This is attributed to the fact that this step needs to be repeated several times to ensure the mAbs can capture sufficient EVs (~240 min).
3.7. Ultrafiltration
Semi-permeable membranes (ranging from 3 kDa to 100 kDa) are adapted for sEV fractionation within filtration-based isolation; the membrane function is determined by its pore size. However, sEVs cannot be fractionated according to their biogenesis or biomarkers, but it is normally used to concentrate sEVs. It is still an efficient way to eliminate the minimal sample volume (~130 min) with a simple procedure, and has been proven to yield higher recovery of sEVs than ultracentrifugation [
49]. However, ultrafiltration might lead to low EV protein, but a rather higher concentration of non-EV proteins (i.e., albumin).
3.8. Current Isolation Challenge
As mentioned previously, the current challenges of sEV isolation include time-consuming procedures, impurities, insufficient EV yield, and low scalability [
50]. Although many researchers have investigated combinations of these isolation methods, an urgent demand has arisen to investigate high-yielding and time-effective isolation protocols. Currently, there is no optimal sEV isolation method; however, a combination of ultracentrifugation, SEC, and ultrafiltration has been used for the pure sEV population, which is a critical factor for downstream therapeutic applications.
4. sEV Isolation and Characterization Methods for Periodontal (and Dental Pulp) Cells
To date, there are no standardized protocols for sEV isolation and characterization. From the 33 studies that are reported in this review, we have summarized periodontal (dental pulp) cell-derived sEV isolation and characterisation methods [
51,
52,
53,
54,
55,
56,
57,
58,
59,
60,
61,
62,
63,
64,
65,
66,
67,
68,
69,
70,
71,
72,
73,
74,
75,
76,
77,
78,
79,
80,
81,
82,
83]. Various isolation methods have been used for periodontal (dental pulp) cells sEVs, including ultracentrifugation (UC), precipitation-based methods, and ultrafiltration (
Figure 3a). Regarding EV characterization, the latest MISEV 2018 guidelines [
6] suggest that all EV researchers should characterize sEV from at least three different aspects, such as EV particle numbers, EV morphology, and EV-enriched protein markers. However, most of the current studies did not follow the MISEV guidelines, and this requires additional attention for all EV research.
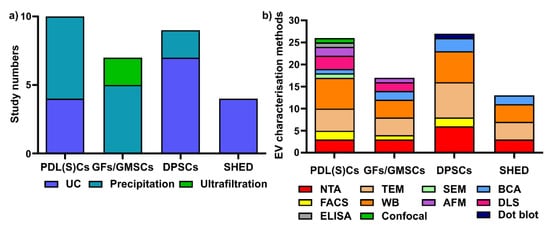
Figure 3. Various sEV isolation methods (a) and characterization methods (b) are used for periodontal (dental pulp) cells. UC: ultracentrifugation; NTA: nanoparticle tracking analysis; TEM: transmission electron microscopy; WB: Western blot; SEM: scanning electron microscopy; AFM: atomic force microscopy; BCA: bicinchoninic acid assay; DLS: dynamic light scattering; ELISA: enzyme-linked immunosorbent assay; confocal: confocal microscopy.
Different sEV isolation methods have been utilized for various cells (Figure 3a), with precipitation and ultracentrifugation methods being the two most commonly used techniques. In PDL(S)C-derived sEV isolation (10 studies), the precipitation-based method (i.e., a commercial ExoQuick kit) is the most commonly used (n = 6, 60%), followed by ultracentrifugation (n = 4, 40%). Among six studies in GFs/GMSC-derived sEVs, the precipitation-based method (n = 4, 66.7%) and ultrafiltration (n = 2, 33.3%) were used for GFs/GMMSCs–sEV isolation. Regarding DPSC-derived sEV, most researchers selected ultracentrifugation (n = 7, 77.8%), with one study using the precipitation-based method (n = 2, 22.2%). For SHED–sEVs, all of the studies (n = 4, 100%) used the ultracentrifugation method to isolate sEVs from SHED.
Concerning EV characterisation [
6], NTA and DLS are common methods to quantify EV particle number, size, and distribution; TEM, SEM, and AFM can be used for EV morphology and size; BCA is for EV protein quantification; and WB is to determine EV-enriched protein markers. We have summarized the various EV characterisation methods for periodontal cell-derived sEVs (
Figure 3b). For PDL(S)Cs—sEV (10 studies included in this review), WB is the most commonly used characterization method (
n = 7 studies), followed by TEM (
n = 5), NTA (
n = 3), AFM (
n = 2), flow cytometry (
n = 2), SEM (
n = 1), BCA assay (
n = 1), ELISA (
n = 1), and confocal microscopy (
n = 1). In GFs/GMMSCs–sEVs, WB (
n = 4) was utilized to detect CD9, CD63, and TSG101, as well as TEM (
n = 4), NTA (
n = 3), DLS (
n = 2), BCA assay (
n = 2), AFM (
n = 1), and FACS (
n = 1). The characterisation of DPSC–EVs are mostly performed using WB (
n = 7), TEM (
n = 8), NTA (
n = 6), BCA assay (
n = 3), FACS (
n = 2), and dot blot (
n = 1). For the characterization of SHED–sEVs (4 studies), TEM and WB (
n = 4) are most commonly applied; NTA (
n = 3) and BCA (
n = 2) were also used for OBs–sEVs.
In summary, ultracentrifugation and precipitation-based methods are the two most common methods used for periodontal (dental pulp) cells sEV isolation. WB, TEM and NTA are the most common methods for periodontal (dental pulp) cell-derived sEVs characterisation.
This entry is adapted from the peer-reviewed paper 10.3390/nano11071858