Inflammatory bowel disease (IBD) is a complex inflammatory chronic and pathological condition that includes Crohn’s disease (CD) and Ulcerative colitis (UC). CD primarily affects the small and large intestine whereas the prime targets for UC are colon and rectum. Immune sentinel subsets of CD4+ T cells such as Th (T helper cells)-1, Th2, Th17, and regulatory T cells (Tregs) play a crucial role in the pathogenesis of IBD. Immunological balance between effector Th cells and Trges is essential for maintaining immune-homeostasis. Immunoregulatory Trges are characterized by the expression of transcription factor Forkhead box P3 (Foxp3), and surface marker CD25, and are functionally immunosuppressive & important for immune tolerance. Therapeutic arrangement based on Tregs is important to address the systemic inflammatory and autoimmune diseases such as IBD and rheumatoid arthritis.
- inflammatory bowels disease
- Crohn’s disease
- ulcerative colitis
- human immune system
- regulatory T cells
1. IBD Pathogenesis
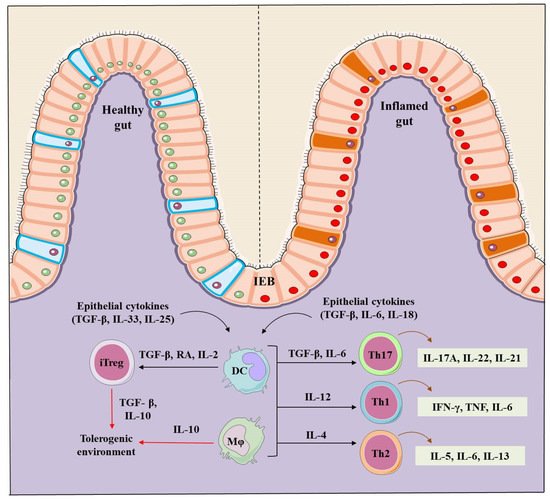
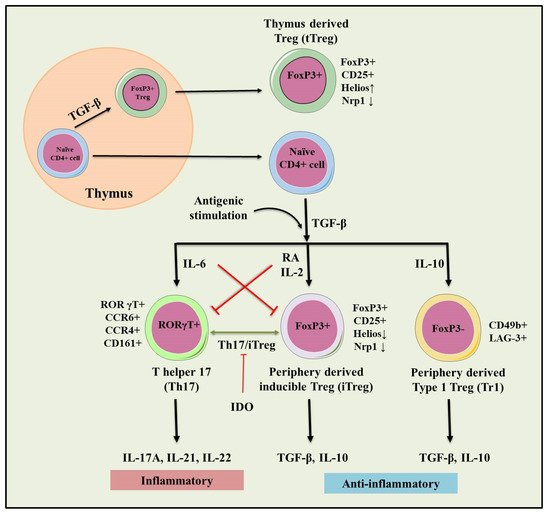
2. Regulatory T Cells (Tregs)
2.1. tTregs and pTre
2.2. Type 1 T regulatory (Tr1) Cells
3. Role of Tregs in IBD
4. Therapeutic Role of Tregs in IBD
This entry is adapted from the peer-reviewed paper 10.3390/cells10081847