New SERDs are currently under development capable of reducing ERα protein expression and blocking estrogen-dependent and independent ER signaling. SERDs are therefore considered a significant therapeutic approach to treat ER+ BC in both early stage and more advanced drug-resistant cases.
- SERD
- breast cancer
- endocrine therapy
- hormone therapy
- luminal breast cancer
1. Introduction
Breast cancer (BC) is a heterogeneous disease comprising different subtypes, which can be identified through molecular biomarkers that also act as predictive factors. Luminal BC is characterized by the expression of estrogen receptor-positive (ER+) and/or progesterone receptor-positive (PR+), HER2-positive BC is defined by overexpression of human epidermal growth factor 2 (HER2) oncogene and conversely, triple-negative BC is characterized by lack of expression of ER/PR and HER2.
Among these, luminal is the most common BC subtype. In the case of metastatic breast cancer, luminal subtype accounts for more than sixty-five percent of all cases. Recommended treatment is endocrine-based systemic therapy, since multiple publications and consensus recommendations conclude that chemotherapy would not be the best option for endocrine sensitive disease, except in situations such as visceral crisis [1].
Endocrine therapy (ET) comprises different strategies as suppression of estrogen production or directly targeting the estrogen receptor (ER). For example, aromatase inhibitors (AIs) (letrozole, anastrozole and exemestane) are potent inhibitors of the aromatase enzyme, which catalyzes the last step in estrogen biosynthesis. These agents decrease estrogen production by blocking androgen conversion to estrogens [2][3].
Direct targeting of ERα is achieved by selective estrogen receptor modulator (SERM) (e.g., tamoxifen) and selective estrogen receptor degrader (SERD) (e.g., fulvestrant). SERMs compete with estrogen for ER binding and show mixed agonist/antagonist capabilities in a tissue-specific fashion. Meanwhile, SERDs create an unstable protein complex that induces ER protein degradation via proteasome [4]. Fulvestrant is a first-generation SERD approved by the FDA in 2007 for treatment of metastatic luminal BC in postmenopausal patients following progression on prior ET with AI or tamoxifen [5][6].
2. Antiestrogen Therapy: Basic Concepts Regarding Old and New Agents
Two major isoforms of the estrogen receptor have been identified, ERα and ERβ: however, the role of ERβ in cancer remains unclear [7][8]. The two isoforms are encoded by two genes located on different chromosomes ( ESR1 on chromosome 6 and ESR2 on chromosome 14), and regulate different specific genes [9][10]. Both isoforms are structurally organized in six different functional domains (A to F). The receptor contains two activation functions (AF) regions (AF-1: domains A/B and AF-2 domains E/F) , responsible for the transcriptional activation of the receptor. C domain is the DNA-binding region, while D domain is a flexible hinge region containing the nuclear localization signal and links the C to E domain. Finally, E domain harbors the hormone-binding site [11].
While ESR1 alterations, such as amplifications, can be identified in up to 30% of ER+ BC patients [12][13], it is still uncertain whether this alteration has clinical significance in terms of ET resistance: while some studies have found that ESR1 amplifications were associated with improved disease-free survival [14][15] several others studies report an association between ESR1 amplifications and tamoxifen resistance [16][17].
Similarly, clinical outcomes for ESR1 fusions require further investigation and efforts, since to this date conclusion cannot be drawn regarding their implications [18]. Fusions and rearrangements are estimated to have an incidence of 1%, mainly involving the first two noncoding exons of ESR1 binding to various C-terminal sequences from the coiled-coil domain-containing 170 genes (CCDC170) ( ESR1 -e2 > CCDC170), consequently conferring endocrine resistance to tamoxifen [19].
A retrospective analysis of the SoFEA phase III trial showed that median PFS in fulvestrant-containing regimens was significantly better than those treated with exemestane (HR = 0.52; 95% CI 0.30–0.92; p = 0.02) for metastatic BC (MBC) and ESR1 mutations [20]. This data may suggest that fulvestrant could be a potentially more adequate ET for ESR1 mutated patients. Conversely, ESR1 Y735S mutations may reflect higher resistance to fulvestrant [21][22].
3. Novel Strategies
PROTACs are heterobifunctional molecules made up of a ligand for ER (target protein) and another ligand, serving as the E3 ubiquitin ligase complex substrate. Once PROTACs bind to ER, recruit the E3 ubiquitin ligase complex, leading to a polyubiquitilation of ER ending on a proteasomal degradation [23]. PROTACs produce a rapid and complete elimination of intracellular receptor and inhibition of ER signaling [24][25]. PROTACs action is pure antagonism of ER realized by elimination of the receptor, rather than conformational changes of ER to block transcriptional activation. Only a transient binding event is required for degradation, and the PROTAC molecules can cycle through multiple rounds of activity, removing substoichiometric quantities of proteins. ( Figure 4 )
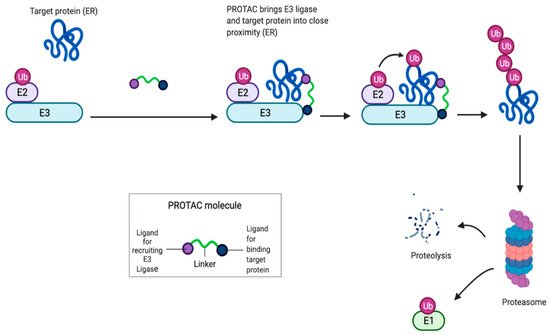
The rapid progress in ER PROTACs development in preclinical studies lead to a first-in-class, orally bioavailable ER degrading agent, ARV-471, which entered clinical trials in 2019 (NCT04072952) ( Table 1 ).
| AGENT | TREATMENT | DISEASE SETTING | PHASE | NAME (INDICATOR) |
|---|---|---|---|---|
| SERMs | ||||
| LASOFOXIFENE | vs. FULVESTRANT +ABEMACICLIB |
Previously treated advanced/metastatic disease with ESR1 mutations Previously treated advanced/metastatic disease with ESR1 mutations |
2 2 |
ELAINE: NCT03781063 ELAINE 2: NCT04432454 |
| BAZEDOXIFENE | +PALBOCICLIB | Previously treated advanced/metastatic | 1/2 | NCT02448771 |
| SERDs | ||||
| LSZ102 | SINGLE AGENT/+ RIBOCICLIB/+ ALPELISIB |
Previously treated advanced/metastatic | 1/1b | NCT02734615 |
| G1T48 (RINTODESTRANT) |
SINGLE AGENT+/− PALBOCICLIB |
Previously treated advanced/metastatic | 1 | NCT03455270 |
| RAD1901 (ELACESTRANT) |
vs. SOC (standard of care) | Previously treated advanced/metastatic | 3 | EMERALD: NCT03778931 |
| GDC-9545 (GIREDESTRANT) |
GDC-9545 vs. LETROZOLE + PALBOCICLIB GDC-9545 vs. ANASTROZOLE + PALBOCICLIB vs. physician’s choice of endocrine therapy +/− PALBOCICLIB and LHRH agonist Monotherapy |
Advanced/metastatic Treatment-naïve early breast cancer (window-of-opportunity -> neoadjuvant) Previously treated advanced/metastatic Advanced/metastatic Treatment-naïve early breast cancer (window-of-opportunity) |
3 2 2 1 1 |
persevERA: NCT04546009 coopERA: NCT04436744 acelERA: NCT04576455 NCT03332797 NCT03916744 |
| SAR439859 (AMCENESTRANT) |
SAR439859 vs. LETROZOLE + PALBOCICLIB vs. physician’s choice of endocrine therapy vs. LETROZOLE +/− PALBOCICLIB OR ALPELISIB |
Advanced/metastatic Previously treated advanced/metastatic Newly diagnosed advanced/metastatic Advanced/metastatic |
3 2 2 1/2 |
AMEERA-5: NCT04478266 AMEERA-3: NCT04059484 AMEERA-4: NCT04191382 AMEERA-1 NCT03284957 |
| AZD9833 (CAMIZESTRANT) |
AZD9833 vs. LETROZOLE + PALBOCICLIB MONOTHERAPY vs. FULVESTRANT +/− PALBOCICLIB, EVEROLIMUS OR ABEMACICLIB |
Treatment-naïve advanced/metastatic Neoadjuvant treatment Previously treated advanced/metastatic Previously treated advanced/metastatic |
3 2 2 1 |
SERENA-4: NCT04711252 SERENA-3: NCT04588298 SERENA-2: NCT04214288 SERENA-1: NCT03616587 |
| LY3484356 | +/− other anticancer therapies MONOTHERAPY |
Advanced/metastatic Neoadjuvant treatment |
1 1 |
EMBER: NCT04188548 EMBER 2: NCT04647487 |
| Zn-c5 | SINGLE AGENT +/− PALBOCICLIB |
Previously treated advanced/metastatic | 1/2 | NCT03560531 |
| D-0502 | SINGLE AGENT +/− PALBOCICLIB |
Previously treated advanced/metastatic | 1 | NCT03471663 |
| NOVEL THERAPIES |
||||
| ARV-471 (PROTAC) | +/− PALBOCICLIB | Previously treated advanced/metastatic | 1/2 | NCT04072952 |
| H3B-5942 (SERCA) | MONOTHERAPY + PALBOCICLIB |
Previously treated advanced/metastatic Previously treated advanced/metastatic |
1/2 1 |
NCT03250676 NCT04288089 |
Abbreviations: ORR: overall response rate; CBR: clinical benefit rate; PFS: progression-free survival; AEs: adverse events; AAT: aspartate aminotransferase; DLT: dose-limiting toxicities; SD: stable disease.
ARV-471 is a PROTAC in which E2 is linked to a small-molecule ubiquitin E3 ligase–binding moiety, facilitating the interaction between the ER and an E3 ligase complex that will tag the ER for degradation by the ubiquitin-proteasome system [26].
A total of 130 patients were enrolled (47 in the phase I part and 83 in the phase II part of the trial) and 105 (58% ER1-mutated) were response-evaluable. The phase I evaluated once daily doses from 100 to 600 mg and the dose of 450 mg was selected as the RP2D. Median age was 62 years and in MBC, the median number of prior therapies was three. Prior CDK4/6i, fulvestrant, and chemotherapy were received by 87%, 71%, and 54% of the patients, respectively. Regarding toxicities, grade 2 or higher adverse events reported in ≥10% were anemia (20%), fatigue (16%), nausea (14%), diarrhea (11%) and AST increase (11%). In the response-evaluable group, 13 confirmed PR (12%). SD and CBR (≥23 weeks) were 45% and 33% respectively at 450 mg. Three PRs (25%) and four SDs were observed in 12 patients in whom clonal ESR1 Y537S was present. Median PFS in all patients was 3.7 months and in ESR1 -mutated patients (Y537S) was 7.3 months [27] ( Table 2 ).
| LSZ102 | Phase I/Ib (NCT02734615) (65) Arm A: Monotherapy Arm B: Combination with Arm B: Combination with Alpelisib |
Arm A (n: 78): ORR (1.3%), CBR (9.1%), PFS (1.8 m) Arm B (n: 76): ORR (15.8%), CBR (35.5%), PFS (6.2 m) Arm C (n: 39): ORR (5.4%), CBR (18.9%), PFS 3.5m |
Arm A, B, C: Grade 3/4: Nausea (3.1%) and Diarrhea (6.7%) Arm B (Grade 3 AEs): Neutropenia (13.2%) and Increased AAT (3.9%) Arm C (Grade 3 AEs): Hyperglycemia (10%), Skin Rashes (15.4%) |
|---|---|---|---|
| RAD1901 (ELACESTRANT) |
Phase I (NCT02338349) (73) |
N: 50 (dose-escalation) ORR 19.4% N: 47 (dose expansion) CBR 42.6% |
No DLTs Grade 1/2: nausea (33.3%), increased triglycerides (25%), decreased blood phosphorus (25%) |
| GDC-9545 (GIREDESTRANT) |
Phase Ib/II (NCT03332797) (88) Dose expansion: Cohort A: monotherapy Cohort B: combination with palbociclib |
N: 88 Cohort A, n: 39 ORR (13%), PFS (7.8 m) Cohort B, n:43 ORR (33%), PFS (9.3 m) |
Cohort A: Grade 1/2 fatigue, arthralgia. Grade 3: Fatigue (1), diarrhea (1), transaminase increased (1) Cohort B: Grade 1/2: neutropenia, fatigue, bradycardia, diarrhea, constipation, dizziness, nauseas, anemia, asthenia, pruritus and visual impairment. Grade 3: neutropenia (50%) |
| SAR439859 (AMCENESTRANT) | Phase I: AMEERA-1 (NCT03284957). (84) Monotherapy dose-escalation (Part A) |
Part A: n: 59 ORR 8.5%, CBR (33.5%) |
Part A: hot flushes (16.1%), constipation (9.7%), arthralgia (9.7%), decreased appetite (8.1%), vomiting (8.1%), diarrhea (8.1%), nausea (8.1%), and fatigue (6.5%) |
| AZD9833 (CAMIZESTRANT) |
Phase I: SERENA-1 (NCT03616587) (87) Part A and B: monotherapy Part C and D: Combination with palbociclib |
Part A and B, n: 98 ORR (10%), CDR (35.3%), PFS (5.4m) Part B and C, n: 48 ORR (6.3%), CBR (50%) |
5 dose-limiting toxicities (3 for monotherapy and 2 for combination therapy) Monotherapy (≥Grade 2 instances of AZD9833-related adverse events): fatigue (9%), bradicardia (3.1%), nausea (3%), visual disturbances (1.1%) Combination: grade 1–2: anemia, fatigue, lymphopenia, nausea, neutropenia, thrombocytopenia, and reduced white blood cell count |
| LY3484356 | Phase I/Ib (89) EMBER (NCT04188548) |
N: 28 | Grade 1–2: nausea (32%), fatigue (25%), and diarrhea (18%) |
| H3B-5942 (SERCA) | Phase 1/2 (NCT03250676) (97) |
N: 130 (phase I n: 47/phase II n: 83) PR (12%). SD (45%) and CBR (33%), PFS (3.7m) |
Grade 2 or higher adverse: anemia (20%), fatigue (16%), nausea (14%), diarrhea (11%) and AST increase (11%). |
4. Summary
ER is involved in the initiation of BC tumorigenesis and in the progression of disease after ET. Targeting, modulating, and degrading ER is the goal of new drugs development, including ESR1 gene mutations identified after ET. Fulvestrant is the only approved SERD and can be used in first-line treatment or after AI or tamoxifen progression. Overcoming fulvestrant’s limitations, new SERDs are currently in early-phase clinical development and some of them in phase III clinical trials. New SERDs have demonstrated improved pharmacokinetic and bioavailability compared to fulvestrant in preclinical and early studies, with a potentially higher clinical benefit rate. In this line, PROTACs and SERCAs open new paths to degrade ER, and are still in early clinical studies.
All the currently available results need to be confirmed in phase III clinical trials with larger patient population, exploring the activity of ET plus CDK4/6i combination progression disease setting.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22157812
References
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. Ohno 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649.
- Santen, R.J.; Brodie, H.; Simpson, E.R.; Siiteri, P.K.; Brodie, A. History of aromatase: Saga of an important biological mediator and therapeutic target. Endocr. Rev. 2009, 30, 343–375.
- Dixon, J.M. Endocrine Resistance in Breast Cancer. N. J. Sci. 2014, 2014, 1–27.
- Liu, S.; Han, S.J.; Smith, C.L. Cooperative activation of gene expression by agonists and antagonists mediated by estrogen receptor heteroligand dimer complexes. Mol. Pharmacol. 2013, 83, 1066–1077.
- Di Leo, A.; Jerusalem, G.; Petruzelka, L.; Torres, R.; Bondarenko, I.; Khasanov, R.; Verhoeven, D.; Pedrini, J.L.; Smirnova, I.; Lichinitser, M.R.; et al. Results of the CONFIRM Phase III Trial Comparing Fulvestrant 250 mg With Fulvestrant 500 mg in Postmenopausal Women with Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2010, 28, 4594–4600.
- Robertson, J.F.; Lindemann, J.; Garnett, S.; Anderson, E.; Nicholson, R.I.; Kuter, I.; Gee, J.M. A Good Drug Made Better: The Fulvestrant Dose-Response Story. Clin. Breast Cancer 2014, 14, 381–389.
- Speirs, V. Oestrogen receptor beta in breast cancer: Good, bad or still too early to tell? J. Pathol. 2002, 197, 143–147.
- Omoto, Y.; Iwase, H. Clinical significance of estrogen receptor beta in breast and prostate cancer from biological aspects. Cancer Sci. 2015, 106, 337–343.
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjöld, M.; Gustafsson, J.-A. Human Estrogen Receptor β-Gene Structure, Chromosomal Localization, and Expression Pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265.
- Menasce, L.P.; White, G.R.M.; Harrison, C.J.; Boyle, J.M. Localization of the estrogen receptor locus (ESR) to chromosome 6q25.1 by FISH and a simple post-FISH banding technique. Genomics 1993, 17, 263–265.
- Lu, Y.; Gutgesell, L.M.; Xiong, R.; Zhao, J.; Li, Y.; Rosales, C.I.; Hollas, M.; Shen, Z.; Gordon-Blake, J.; Dye, K.; et al. Design and Synthesis of Basic. Selective Estrogen Receptor Degraders for Endocrine Therapy Resistant Breast Cancer. J. Med. Chem. 2019, 62, 11301–11323.
- Basudan, A.; Priedigkeit, N.; Hartmaier, R.J.; Sokol, E.S.; Bahreini, A.; Watters, R.J.; Boisen, M.M.; Bhargava, R.; Weiss, K.R.; Karsten, M.M.; et al. Frequent ESR1 and CDK Pathway Copy-Number Alterations in Metastatic Breast Cancer. Mol. Cancer Res. 2018, 17, 457–468.
- Brown, L.; Hoog, J.; Chin, S.-F.; Tao, Y.; Zayed, A.A.; Chin, K.; Teschendorff, A.E.; Quackenbush, J.F.; Marioni, J.C.; Leung, S.; et al. ESR1 gene amplification in breast cancer: A common phenomenon? Nat. Genet. 2008, 40, 806–807.
- Tomita, S.; Zhang, Z.; Nakano, M.; Ibusuki, M.; Kawazoe, T.; Yamamoto, Y.; Iwase, H. Estrogen receptor α geneESR1amplification may predict endocrine therapy responsiveness in breast cancer patients. Cancer Sci. 2009, 100, 1012–1017.
- Holst, F.; Singer, C.F. ESR1-Amplification-Associated Estrogen Receptor α Activity in Breast Cancer. Trends Endocrinol. Metabolites 2016, 27, 751–752.
- Nielsen, K.V.; Ejlertsen, B.; Müller, S.; Møller, S.; Rasmussen, B.B.; Balslev, E.; Lænkholm, A.-V.; Christiansen, P.; Mouridsen, H.T. Amplification of ESR1 may predict resistance to adjuvant tamoxifen in postmenopausal patients with hormone receptor positive breast cancer. Breast Cancer Res. Treat. 2010, 127, 345–355.
- Markiewicz, A.; Welnicka-Jaskiewicz, M.; Skokowski, J.; Jaśkiewicz, J.; Szade, J.; Jassem, J.; Zaczek, A.J. Prognostic Significance of ESR1 Amplification and ESR1 PvuII, CYP2C19*2, UGT2B15*2 Polymorphisms in Breast Cancer Patients. PLoS ONE 2013, 8, e72219.
- Lee, N.; Park, M.-J.; Song, W.; Jeon, K.; Jeong, S. Currently Applied Molecular Assays for Identifying ESR1 Mutations in Patients with Advanced Breast Cancer. Int. J. Mol. Sci. 2020, 21, 8807.
- Veeraraghavan, J.; Tan, Y.; Cao, X.-X.; Kim, J.A.; Wang, X.; Chamness, G.C.; Maiti, S.N.; Cooper, L.J.N.; Edwards, D.P.; Contreras, A.; et al. Recurrent ESR1–CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat. Commun. 2014, 5, 1–12.
- Fribbens, C.; O’Leary, B.; Kilburn, L.; Hrebien, S.; Garcia-Murillas, I.; Beaney, M.; Cristofanilli, M.; Andre, F.; Loi, S.; Loibl, S.; et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor–Positive Advanced Breast Cancer. J. Clin. Oncol. 2016, 34, 2961–2968.
- De Santo, I.; McCartney, A.; Migliaccio, I.; Di Leo, A.; Malorni, L. The emerging role of ESR1 mutations in luminal breast cancer as a prognostic and predictive biomarker of response to endocrine therapy. Cancers 2019, 11, 1894.
- Toy, W.; Weir, H.; Razavi, P. Activating ESR1 mutations differentially impact the efficacy of ER antagonists. Cancer Discov. 2017, 7, 277–287.
- Dougan, D.A.; Micevski, D.; Truscott, K.N. The N-end rule pathway: From recognition by N-recognins, to destruction by AAA +proteases. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 83–91.
- Flanagan, J.; Qian, Y.; Gough, S.; Andreoli, M.; Bookbinder, M.; Cadelina, G.; Bradley, J.; Rousseau, E.; Willard, R.; Pizzano, J.; et al. Abstract P5-04-18: ARV-471, an oral estrogen receptor PROTAC degrader for breast cancer. Poster Sess. Abstr. 2019, 79.
- Hu, J.; Hu, B.; Wang, M.; Xu, F.; Miao, B.; Yang, C.Y.; Wang, M.; Liu, Z.; Hayes, D.F.; Chinnaswamy, K.; et al. Discovery of ERD-308 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Estrogen Receptor (ER). J. Med. Chem. 2019, 62, 1420–1442.
- Gao, H.; Sun, X.; Rao, Y. PROTAC technology: Opportunities and challenges. ACS Med. Chem Lett. 2020, 11, 237–240.
- Hamilton, E.P.; Wang, J.S.; Pluard, T.; Johnston, S.; Morikawa, A.A.; Dees, C.E.; Jones, R.H.; Haley, B.; Armstrong, A.; Cohen, A.L.; et al. Abstract PD8-06: Phase I/II trial of H3B-6545, a novel selective estrogen receptor covalent antagonist (SERCA), in estrogen receptor positive (ER+), human epidermal growth factor receptor 2 negative (HER2-) advanced breast cancer. In Proceedings of the 2020 San Antonio Breast Cancer Symposium, Virtual Conference, 8–11 December 2020. Abstract 1142.