Neutrophils are first-line responders to infections and are recruited to target tissues through the action of chemoattractant molecules, such as chemokines. Neutrophils are crucial for the control of bacterial and fungal infections, but their role in the context of viral infections has been understudied. Flaviviruses are important human viral pathogens transmitted by arthropods. Infection with a flavivirus may result in a variety of complex disease manifestations, including hemorrhagic fever, encephalitis or congenital malformations. Our understanding of flaviviral diseases is incomplete, and so is the role of neutrophils in such diseases. Here we present a comprehensive overview on the participation of neutrophils in severe disease forms evolving from flavivirus infection, focusing on the role of chemokines and their receptors as main drivers of neutrophil function. Neutrophil activation during viral infection was shown to interfere in viral replication through effector functions, but the resulting inflammation is significant and may be detrimental to the host. For congenital infections in humans, neutrophil recruitment mediated by CXCL8 would be catastrophic.
- neutrophils
- flavivirus
- encephalitis
- hemorrhagic fever
- pregnancy
1. Flaviviruses
2. Participation of Neutrophils and Neutrophil-Associated Molecules in Flaviviral Diseases
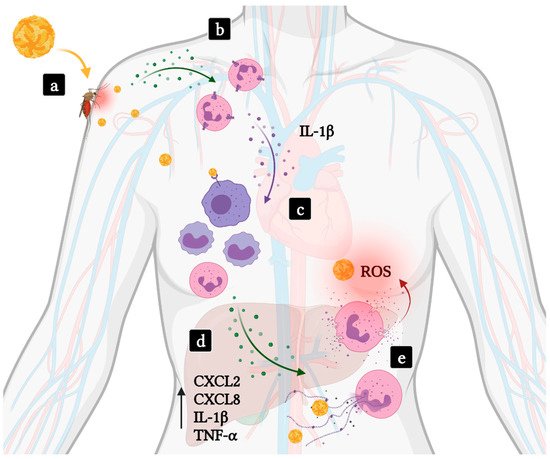
2.1. Hemorrhagic Fevers
2.2. Encephalitis
2.2.1. SLEV
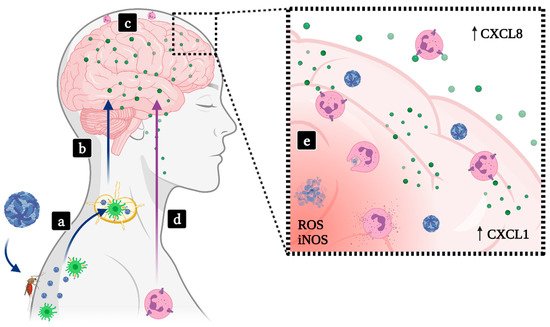
2.2.2. JEV
2.2.3. WNV
2.2.4. MVEV
2.2.5. TBEV
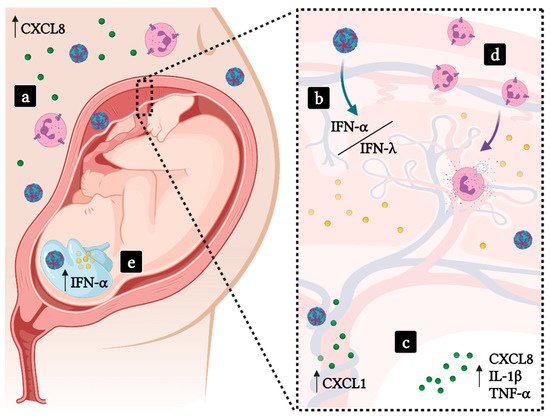
2.3. Implications in Pregnancy
This entry is adapted from the peer-reviewed paper 10.3390/life11070717
References
- Marques, R.E.; Guabiraba, R.; Cisalpino, D.; Teixeira, M.M.; Souza, D.G. Dengue. Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2014; Volume 6, pp. 1–104.
- Rothman, A.L. Immunity to Dengue Virus: A Tale of Original Antigenic Sin and Tropical Cytokine Storms. Nat. Rev. Immunol. 2011, 11, 532–543.
- Rosa, R.; Costa, E.A.; Marques, R.E.; Oliveira, T.S.; Furtini, R.; Bomfim, M.R.Q.; Teixeira, M.M.; Paixão, T.A.; Santos, R.L. Isolation of Saint Louis Encephalitis Virus from a Horse with Neurological Disease in Brazil. PLoS Negl. Trop. Dis. 2013, 7, e2537.
- Ribeiro, L.S.; Marques, R.E.; de Jesus, A.M.R.; de Almeida, R.P.; Teixeira, M.M. Zika Crisis in Brazil: Challenges in Research and Development. Curr. Opin. Virol. 2016, 18, 76–81.
- Zuchi, N.; da Silva Heinen, L.B.; dos Santos, M.A.M.; Pereira, F.C.; Slhessarenko, R.D. Molecular Detection of Mayaro Virus during a Dengue Outbreak in the State of Mato Grosso, Central-West Brazil. Mem. Inst. Oswaldo Cruz 2014, 109, 820–823.
- Tamba, M.; Bonilauri, P.; Bellini, R.; Calzolari, M.; Albieri, A.; Sambri, V.; Dottori, M.; Angelini, P. Detection of Usutu Virus within a West Nile Virus Surveillance Program in Northern Italy. Vector Borne Zoonotic Dis. 2011, 11, 551–557.
- Pierson, T.C.; Diamond, M.S. The Continued Threat of Emerging Flaviviruses. Nat. Microbiol. 2020, 5, 796–812.
- Rocha, R.F.; Del Sarto, J.L.; Marques, R.E.; Costa, V.V.; Teixeira, M.M. Host Target-Based Approaches against Arboviral Diseases. Biol. Chem. 2018, 399, 203–217.
- Viral Hemorrhagic Fevers (VHFs). Available online: (accessed on 17 June 2021).
- Bray, M. Pathogenesis of Viral Hemorrhagic Fever. Curr. Opin. Immunol. 2005, 17, 399–403.
- LeDuc, J.W. Epidemiology of Hemorrhagic Fever Viruses. Rev. Infect. Dis. 1989, 11 (Suppl. 4), S730–S735.
- Guerrero, D.; Cantaert, T.; Missé, D. Aedes Mosquito Salivary Components and Their Effect on the Immune Response to Arboviruses. Front. Cell. Infect. Microbiol. 2020, 10, 407.
- Pingen, M.; Bryden, S.R.; Pondeville, E.; Schnettler, E.; Kohl, A.; Merits, A.; Fazakerley, J.K.; Graham, G.J.; McKimmie, C.S. Host Inflammatory Response to Mosquito Bites Enhances the Severity of Arbovirus Infection. Immunity 2016, 44, 1455–1469.
- Stegelmeier, A.A.; van Vloten, J.P.; Mould, R.C.; Klafuric, E.M.; Minott, J.A.; Wootton, S.K.; Bridle, B.W.; Karimi, K. Myeloid Cells during Viral Infections and Inflammation. Viruses 2019, 11, 168.
- Findlay, J.S.; Ulaeto, D.; D’Elia, R.V. Cytokines and Viral Hemorrhagic Fever: Potential for Therapeutic Intervention. Future Virol. 2015, 10, 547–557.
- Messaoudi, I.; Basler, C.F. Immunological Features Underlying Viral Hemorrhagic Fevers. Curr. Opin. Immunol. 2015, 36, 38–46.
- Schulz, C.; Gabriel, G.; von Köckritz-Blickwede, M. Detrimental Role of Neutrophil Extracellular Traps during Dengue Virus Infection. Trends Immunol. 2020, 41, 3–6.
- Opasawatchai, A.; Amornsupawat, P.; Jiravejchakul, N.; Chan-In, W.; Spoerk, N.J.; Manopwisedjaroen, K.; Singhasivanon, P.; Yingtaweesak, T.; Suraamornkul, S.; Mongkolsapaya, J.; et al. Neutrophil Activation and Early Features of NET Formation Are Associated with Dengue Virus Infection in Human. Front. Immunol. 2018, 9, 3007.
- Hoang, L.T.; Lynn, D.J.; Henn, M.; Birren, B.W.; Lennon, N.J.; Le, P.T.; Duong, K.T.H.; Nguyen, T.T.H.; Mai, L.N.; Farrar, J.J.; et al. The Early Whole-Blood Transcriptional Signature of Dengue Virus and Features Associated with Progression to Dengue Shock Syndrome in Vietnamese Children and Young Adults. J. Virol. 2010, 84, 12982–12994.
- Thein, T.-L.; Lye, D.C.; Leo, Y.-S.; Wong, J.G.X.; Hao, Y.; Wilder-Smith, A. Severe Neutropenia in Dengue Patients: Prevalence and Significance. Am. J. Trop. Med. Hyg. 2014, 90, 984–987.
- Turtle, L.; Griffiths, M.J.; Solomon, T. Encephalitis Caused by Flaviviruses. QJM Int. J. Med. 2012, 105, 219–223.
- Meyding-Lamadé, U.; Craemer, E.; Schnitzler, P. Emerging and Re-Emerging Viruses Affecting the Nervous System. Neurol. Res. Pract. 2019, 1, 20.
- Salimi, H.; Cain, M.D.; Klein, R.S. Encephalitic Arboviruses: Emergence, Clinical Presentation, and Neuropathogenesis. Neurotherapeutics 2016, 13, 514–534.
- Moore, S.M. The Current Burden of Japanese Encephalitis and the Estimated Impacts of Vaccination: Combining Estimates of the Spatial Distribution and Transmission Intensity of a Zoonotic Pathogen. medRxiv 2021.
- Marques, R.E.; Del Sarto, J.L.; Rocha, R.P.F.; Gomes, G.F.; Cramer, A.; Rachid, M.A.; Souza, D.G.; Nogueira, M.L.; Teixeira, M.M. Development of a Model of Saint Louis Encephalitis Infection and Disease in Mice. J. Neuroinflamm. 2017, 14, 61.
- Richner, J.M.; Gmyrek, G.B.; Govero, J.; Tu, Y.; van der Windt, G.J.W.; Metcalf, T.U.; Haddad, E.K.; Textor, J.; Miller, M.J.; Diamond, M.S. Age-Dependent Cell Trafficking Defects in Draining Lymph Nodes Impair Adaptive Immunity and Control of West Nile Virus Infection. PLoS Pathog. 2015, 11, e1005027.
- Ryan, E.T.; Hill, D.R.; Solomon, T.; Aronson, N.E.; Endy, T.P. (Eds.) 38—Viral CNS Infections. In Hunter’s Tropical Medicine and Emerging Infectious Diseases, 10th ed.; Elsevier: London, UK, 2020; pp. 382–420. ISBN 9780323555128.
- Rocha, R.F.; Del Sarto, J.L.; Gomes, G.F.; Gonçalves, M.P.; Rachid, M.A.; Smetana, J.H.C.; Souza, D.G.; Teixeira, M.M.; Marques, R.E. Type I Interferons Are Essential While Type II Interferon Is Dispensable for Protection against St. Louis Encephalitis Virus Infection in the Mouse Brain. Virulence 2021, 12, 244–259.
- Redant, V.; Favoreel, H.W.; Dallmeier, K.; Van Campe, W.; De Regge, N. Efficient Control of Japanese Encephalitis Virus in the Central Nervous System of Infected Pigs Occurs in the Absence of a Pronounced Inflammatory Immune Response. J. Neuroinflamm. 2020, 17, 315.
- Bardina, S.V.; Lim, J.K. The Role of Chemokines in the Pathogenesis of Neurotropic Flaviviruses. Immunol. Res. 2012, 54, 121–132.
- Lannes, N.; Summerfield, A.; Filgueira, L. Regulation of Inflammation in Japanese Encephalitis. J. Neuroinflamm. 2017, 14, 158.
- Su, Q.; Xie, Z.-X.; He, F.; Liu, Z.-C.; Song, X.-J.; Zhao, F.-C.; Li, D.; Che, F.-Y. Adults with Severe Japanese Encephalitis: A Retrospective Analysis of 9 Cases in Linyi, China. Neurol. Sci. 2020.
- Singh, S.; Singh, G.; Tiwari, S.; Kumar, A. CCR2 Inhibition Reduces Neurotoxic Microglia Activation Phenotype After Japanese Encephalitis Viral Infection. Front. Cell. Neurosci. 2020, 14, 230.
- Singh, A.; Kulshreshtha, R.; Mathur, A. Secretion of the Chemokine Interleukin-8 during Japanese Encephalitis Virus Infection. J. Med. Microbiol. 2000, 49, 607–612.
- Srivastava, S.; Khanna, N.; Saxena, S.K.; Singh, A.; Mathur, A.; Dhole, T.N. Degradation of Japanese Encephalitis Virus by Neutrophils. Int. J. Exp. Pathol. 1999, 80, 17–24.
- Debiasi, R.L.; Tyler, K.L. West Nile Virus Meningoencephalitis. Nat. Clin. Pract. Neurol. 2006, 2, 264–275.
- Bai, F.; Kong, K.-F.; Dai, J.; Qian, F.; Zhang, L.; Brown, C.R.; Fikrig, E.; Montgomery, R.R. A Paradoxical Role for Neutrophils in the Pathogenesis of West Nile Virus. J. Infect. Dis. 2010, 202, 1804–1812.
- Paul, A.M.; Acharya, D.; Duty, L.; Thompson, E.A.; Le, L.; Stokic, D.S.; Leis, A.A.; Bai, F. Osteopontin Facilitates West Nile Virus Neuroinvasion via Neutrophil “Trojan Horse” Transport. Sci. Rep. 2017, 7, 1–11.
- Macdonald, F. Murray Valley Encephalitis Infection in the Laboratory Mouse. I. Influence of Age on Susceptibility to Infection. Aust. J. Exp. Biol. Med. Sci. 1952, 30, 319–324.
- Andrews, D.M.; Matthews, V.B.; Sammels, L.M.; Carrello, A.C.; McMinn, P.C. The Severity of Murray Valley Encephalitis in Mice Is Linked to Neutrophil Infiltration and Inducible Nitric Oxide Synthase Activity in the Central Nervous System. J. Virol. 1999, 73, 8781–8790.
- McMinn, P.C.; Dalgarno, L.; Weir, R.C. A Comparison of the Spread of Murray Valley Encephalitis Viruses of High or Low Neuroinvasiveness in the Tissues of Swiss Mice after Peripheral Inoculation. Virology 1996, 220, 414–423.
- Matthews, V.; Robertson, T.; Kendrick, T.; Abdo, M.; Papadimitriou, J.; McMinn, P. Morphological Features of Murray Valley Encephalitis Virus Infection in the Central Nervous System of Swiss Mice. Int. J. Exp. Pathol. 2000, 81, 31–40.
- Hombach, J.; Barrett, A.D.T.; Kollaritsch, H. 59—Tickborne Encephalitis Vaccines. In Plotkin’s Vaccines, 7th ed.; Plotkin, S.A., Orenstein, W.A., Offit, P.A., Edwards, K.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1080–1094.e5. ISBN 9780323357616.
- Calvert, A.; Quenby, M.; Heath, P.T. Chapter 9—Vaccination in pregnancy in specific circumstances. In Maternal Immunization; Leuridan, E.E., Nunes, M.C., Jones, C.E., Eds.; Academic Press: Cambridge, MA, USA, 2020; pp. 191–210. ISBN 9780128145821.
- Plekhova, N.G.; Somova, L.M.; Lyapun, I.N.; Krylova, N.V.; Leonova, G.N. Neutrophil Apoptosis Induction by Tick-Borne Encephalitis Virus. Bull. Exp. Biol. Med. 2012, 153, 105–108.
- Thangamani, S.; Hermance, M.E.; Santos, R.I.; Slovak, M.; Heinze, D.; Widen, S.G.; Kazimirova, M. Transcriptional Immunoprofiling at the Tick-Virus-Host Interface during Early Stages of Tick-Borne Encephalitis Virus Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 494.
- Wilson, J.G. Experimental Studies on Congenital Malformations. J. Chronic Dis. 1959, 10, 111–130.
- Barlow, S. Handbook of Teratology, Volume 1: General Principles and Etiology. Biochem. Soc. Trans. 1978, 6, 473–474.
- Alwan, S.; Chambers, C.D. Identifying Human Teratogens: An Update. J. Pediatr. Genet. 2015, 4, 39–41.
- Nausheen, F.; Bashir, S.S.; Atapattu, D.N. Pregnancy Associated Arbovirus Infections. Clin. Microbiol. Infect. 2018, 3, 1–9.
- Coyne, C.B.; Lazear, H.M. Zika Virus—Reigniting the TORCH. Nat. Rev. Microbiol. 2016, 14, 707–715.
- Ander, S.E.; Diamond, M.S.; Coyne, C.B. Immune Responses at the Maternal-Fetal Interface. Sci. Immunol. 2019, 4.
- Mor, G.; Aldo, P.; Alvero, A.B. The Unique Immunological and Microbial Aspects of Pregnancy. Nat. Rev. Immunol. 2017, 17, 469–482.
- Abu-Raya, B.; Michalski, C.; Sadarangani, M.; Lavoie, P.M. Maternal Immunological Adaptation during Normal Pregnancy. Front. Immunol. 2020, 11, 575197.
- Kourtis, A.P.; Read, J.S.; Jamieson, D.J. Pregnancy and Infection. N. Engl. J. Med. 2014, 370, 2211–2218.
- Morelli, S.S.; Mandal, M.; Goldsmith, L.T.; Kashani, B.N.; Ponzio, N.M. The Maternal Immune System during Pregnancy and Its Influence on Fetal Development. Res. Rep. Biol. 2015, 6, 171–189.
- Thomson, A.J.; Telfer, J.F.; Young, A.; Campbell, S.; Stewart, C.J.; Cameron, I.T.; Greer, I.A.; Norman, J.E. Leukocytes Infiltrate the Myometrium during Human Parturition: Further Evidence That Labour Is an Inflammatory Process. Hum. Reprod. 1999, 14, 229–236.
- Gomez-Lopez, N.; Guilbert, L.J.; Olson, D.M. Invasion of the Leukocytes into the Fetal-Maternal Interface during Pregnancy. J. Leukoc. Biol. 2010, 88, 625–633.
- Kruse, A.; Martens, N.; Fernekorn, U.; Hallmann, R.; Butcher, E.C. Alterations in the Expression of Homing-Associated Molecules at the Maternal/fetal Interface during the Course of Pregnancy. Biol. Reprod. 2002, 66, 333–345.
- Sacks, G.P.; Studena, K.; Sargent, K.; Redman, C.W. Normal Pregnancy and Preeclampsia Both Produce Inflammatory Changes in Peripheral Blood Leukocytes Akin to Those of Sepsis. Am. J. Obstet. Gynecol. 1998, 179, 80–86.
- Giaglis, S.; Stoikou, M.; Sur Chowdhury, C.; Schaefer, G.; Grimolizzi, F.; Rossi, S.W.; Hoesli, I.M.; Lapaire, O.; Hasler, P.; Hahn, S. Multimodal Regulation of NET Formation in Pregnancy: Progesterone Antagonizes the Pro-NETotic Effect of Estrogen and G-CSF. Front. Immunol. 2016, 7, 565.
- Wira, C.R.; Fahey, J.V.; Rodriguez-Garcia, M.; Shen, Z.; Patel, M.V. Regulation of Mucosal Immunity in the Female Reproductive Tract: The Role of Sex Hormones in Immune Protection against Sexually Transmitted Pathogens. Am. J. Reprod. Immunol. 2014, 72, 236–258.
- Li, S.; Herrera, G.G.; Tam, K.K.; Lizarraga, J.S.; Beedle, M.-T.; Winuthayanon, W. Estrogen Action in the Epithelial Cells of the Mouse Vagina Regulates Neutrophil Infiltration and Vaginal Tissue Integrity. Sci. Rep. 2018, 8, 11247.
- Aagaard-Tillery, K.M.; Silver, R.; Dalton, J. Immunology of Normal Pregnancy. Semin. Fetal Neonatal Med. 2006, 11, 279–295.
- Shimoya, K.; Matsuzaki, N.; Taniguchi, T.; Kameda, T.; Koyama, M.; Neki, R.; Saji, F.; Tanizawa, O. Human Placenta Constitutively Produces Interleukin-8 during Pregnancy and Enhances Its Production in Intrauterine Infection. Biol. Reprod. 1992, 47, 220–226.
- Gomez-Lopez, N.; Romero, R.; Xu, Y.; Leng, Y.; Garcia-Flores, V.; Miller, D.; Jacques, S.M.; Hassan, S.S.; Faro, J.; Alsamsam, A.; et al. Are Amniotic Fluid Neutrophils in Women with Intraamniotic Infection And/or Inflammation of Fetal or Maternal Origin? Am. J. Obstet. Gynecol. 2017, 217, 693.e1–693.e16.
- Szarka, A.; Rigó, J., Jr.; Lázár, L.; Beko, G.; Molvarec, A. Circulating Cytokines, Chemokines and Adhesion Molecules in Normal Pregnancy and Preeclampsia Determined by Multiplex Suspension Array. BMC Immunol. 2010, 11, 59.
- Gupta, A.K.; Hasler, P.; Holzgreve, W.; Gebhardt, S.; Hahn, S. Induction of Neutrophil Extracellular DNA Lattices by Placental Microparticles and IL-8 and Their Presence in Preeclampsia. Hum. Immunol. 2005, 66, 1146–1154.
- Vokalova, L.; van Breda, S.V.; Ye, X.L.; Huhn, E.A.; Than, N.G.; Hasler, P.; Lapaire, O.; Hoesli, I.; Rossi, S.W.; Hahn, S. Excessive Neutrophil Activity in Gestational Diabetes Mellitus: Could It Contribute to the Development of Preeclampsia? Front. Endocrinol. 2018, 9, 542.
- Staples, J.E.; Monath, T.P. Yellow Fever: 100 Years of Discovery. JAMA 2008, 300, 960–962.
- Machado, C.R.; Machado, E.S.; Rohloff, R.D.; Azevedo, M.; Campos, D.P.; de Oliveira, R.B.; Brasil, P. Is Pregnancy Associated with Severe Dengue? A Review of Data from the Rio de Janeiro Surveillance Information System. PLoS Negl. Trop. Dis. 2013, 7, e2217.
- Tan, P.C.; Soe, M.Z.; Si Lay, K.; Wang, S.M.; Sekaran, S.D.; Omar, S.Z. Dengue Infection and Miscarriage: A Prospective Case Control Study. PLoS Negl. Trop. Dis. 2012, 6, e1637.
- Paixão, E.S.; Teixeira, M.G.; Costa, M.d.C.N.; Rodrigues, L.C. Dengue during Pregnancy and Adverse Fetal Outcomes: A Systematic Review and Meta-Analysis. Lancet Infect. Dis. 2016, 16, 857–865.
- Sirinavin, S.; Nuntnarumit, P.; Supapannachart, S.; Boonkasidecha, S.; Techasaensiri, C.; Yoksarn, S. Vertical Dengue Infection: Case Reports and Review. Pediatr. Infect. Dis. J. 2004, 23, 1042–1047.
- Basurko, C.; Carles, G.; Youssef, M.; Guindi, W.E.L. Maternal and Fetal Consequences of Dengue Fever during Pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 147, 29–32.
- Sicé, A.; Rodallec, C. Manifestations Hémorragiques de: La Fièvre Jaune (typhus Amaril). Répercussions de L’infection Maternelle Sur L’organisme Foetal. Bull. Soc. Pathol. Exot. 1940, 33, 66–69.
- Bentlin, M.R.; de Barros Almeida, R.A.M.; Coelho, K.I.R.; Ribeiro, A.F.; Siciliano, M.M.; Suzuki, A.; Fortaleza, C.M.C.B. Perinatal Transmission of Yellow Fever, Brazil, 2009. Emerg. Infect. Dis. 2011, 17, 1779–1780.
- Burns, K.F. Congenital Japanese B Encephalitis Infection of Swine. Proc. Soc. Exp. Biol. Med. 1950, 75, 621–625.
- Shimizu, T.; Kawakami, Y.; Fukuhara, S.; Matumoto, M. Experimental Stillbirth in Pregnant Swine Infected with Japanese Encephalitis Virus. Jpn. J. Exp. Med. 1954, 24, 363–375.
- Chaturvedi, U.C.; Mathur, A.; Chandra, A.; Das, S.K.; Tandon, H.O.; Singh, U.K. Transplacental Infection with Japanese Encephalitis Virus. J. Infect. Dis. 1980, 141, 712–715.
- Andersen, A.A.; Hanson, R.P. Experimental Transplacental Transmission of St. Louis Encephalitis Virus in Mice. Infect. Immun. 1970, 2, 320–325.
- Charlier, C.; Beaudoin, M.-C.; Couderc, T.; Lortholary, O.; Lecuit, M. Arboviruses and Pregnancy: Maternal, Fetal, and Neonatal Effects. Lancet Child Adolesc. Health 2017, 1, 134–146.
- Centers for Disease Control and Prevention (CDC). Intrauterine West Nile Virus Infection—New York, 2002. MMWR Morb. Mortal. Wkly. Rep. 2002, 51, 1135–1136.
- Alpert, S.G.; Fergerson, J.; Noël, L.P. Intrauterine West Nile Virus: Ocular and Systemic Findings. Am. J. Ophthalmol. 2003, 136, 733–735.
- O’Leary, D.R.; Kuhn, S.; Kniss, K.L.; Hinckley, A.F.; Rasmussen, S.A.; Pape, W.J.; Kightlinger, L.K.; Beecham, B.D.; Miller, T.K.; Neitzel, D.F.; et al. Birth Outcomes Following West Nile Virus Infection of Pregnant Women in the United States: 2003–2004. Pediatrics 2006, 117, e537–e545.
- Platt, D.J.; Smith, A.M.; Arora, N.; Diamond, M.S.; Coyne, C.B.; Miner, J.J. Zika Virus-Related Neurotropic Flaviviruses Infect Human Placental Explants and Cause Fetal Demise in Mice. Sci. Transl. Med. 2018, 10.
- Morrison, T.E.; Diamond, M.S. Animal Models of Zika Virus Infection, Pathogenesis, and Immunity. J. Virol. 2017, 91, e00009-17.
- Brasil, P.; Pereira, J.P., Jr.; Moreira, M.E.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.-A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334.
- Schuler-Faccini, L.; Ribeiro, E.M.; Feitosa, I.M.L.; Horovitz, D.D.G.; Cavalcanti, D.P.; Pessoa, A.; Doriqui, M.J.R.; Neri, J.I.; Monteiro de Pina Neto, J.; Wanderley, H.Y.C.; et al. Possible Association Between Zika Virus Infection and Microcephaly—Brazil, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 59–62.
- Leal, M.C.; Muniz, L.F.; Ferreira, T.S.A.; Santos, C.M.; Almeida, L.C.; Van Der Linden, V.; Ramos, R.C.F.; Rodrigues, L.C.; Neto, S.S.C. Hearing Loss in Infants with Microcephaly and Evidence of Congenital Zika Virus Infection—Brazil, November 2015–May 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 917–919.
- Camacho-Zavala, E.; Santacruz-Tinoco, C.; Muñoz, E.; Chacón-Salinas, R.; Salazar-Sanchez, M.I.; Grajales, C.; González-Ibarra, J.; Borja-Aburto, V.H.; Jaenisch, T.; Gonzalez-Bonilla, C.R. Pregnant Women Infected with Zika Virus Show Higher Viral Load and Immunoregulatory Cytokines Profile with CXCL10 Increase. Viruses 2021, 13, 80.
- Ornelas, A.M.M.; Pezzuto, P.; Silveira, P.P.; Melo, F.O.; Ferreira, T.A.; Oliveira-Szejnfeld, P.S.; Leal, J.I.; Amorim, M.M.R.; Hamilton, S.; Rawlinson, W.D.; et al. Immune Activation in Amniotic Fluid from Zika Virus-Associated Microcephaly. Ann. Neurol. 2017, 81, 152–156.
- Lima, M.C.; de Mendonça, L.R.; Rezende, A.M.; Carrera, R.M.; Aníbal-Silva, C.E.; Demers, M.; D’Aiuto, L.; Wood, J.; Chowdari, K.V.; Griffiths, M.; et al. The Transcriptional and Protein Profile from Human Infected Neuroprogenitor Cells Is Strongly Correlated to Zika Virus Microcephaly Cytokines Phenotype Evidencing a Persistent Inflammation in the CNS. Front. Immunol. 2019, 10, 1928.
- Khaiboullina, S.; Uppal, T.; Kletenkov, K.; St. Jeor, S.C.; Garanina, E.; Rizvanov, A.; Verma, S.C. Transcriptome Profiling Reveals Pro-Inflammatory Cytokines and Matrix Metalloproteinase Activation in Zika Virus Infected Human Umbilical Vein Endothelial Cells. Front. Pharmacol. 2019, 10, 642.
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T.D.A., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712.
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.-E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-Redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890.e6.
- Blazek, K.; Eames, H.L.; Weiss, M.; Byrne, A.J.; Perocheau, D.; Pease, J.E.; Doyle, S.; McCann, F.; Williams, R.O.; Udalova, I.A. IFN-λ Resolves Inflammation via Suppression of Neutrophil Infiltration and IL-1β Production. J. Exp. Med. 2015, 212, 845–853.
- Azamor, T.; Torrentes-Carvalho, A.; Vasconcelos, Z.; Bom, A.P.D.A.; Melgaço, J.G. Innate Immunity Modulation during Zika Virus Infection on Pregnancy: What We Still Need to Know for Medical Sciences Breakthrough. In Cell Interaction; Singh, B., Ed.; IntechOpen: Rijeka, Croatia, 2021.
- Azamor, T.; Cunha, D.P.; da Silva, A.M.V.; de Lima Bezerra, O.C.; Ribeiro-Alves, M.; Calvo, T.L.; de Souza Gomes Kehdy, F.; de Neves Manta, F.S.; Pinto, T.G.; Ferreira, L.P.; et al. Congenital Zika Syndrome Is Associated with Interferon Alfa Receptor 1. bioRxiv 2020, 715862.
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I Interferons Instigate Fetal Demise after Zika Virus Infection. Sci. Immunol. 2018, 3.
- Iwasaki, A.; Pillai, P.S. Innate Immunity to Influenza Virus Infection. Nat. Rev. Immunol. 2014, 14, 315–328.
- Andrade, D.; Kim, M.; Blanco, L.P.; Karumanchi, S.A.; Koo, G.C.; Redecha, P.; Kirou, K.; Alvarez, A.M.; Mulla, M.J.; Crow, M.K.; et al. Interferon-α and Angiogenic Dysregulation in Pregnant Lupus Patients Who Develop Preeclampsia. Arthritis Rheumatol. 2015, 67, 977–987.