The circadian rhythm plays a chief role in the adaptation of all bodily processes to internal and environmental changes on the daily basis. Next to light/dark phases, feeding patterns constitute the most essential element entraining daily oscillations, and therefore, timely and appropriate restrictive diets have a great capacity to restore the circadian rhythm. One of the restrictive nutritional approaches, caloric restriction (CR) achieves stunning results in extending health span and life span via coordinated changes in multiple biological functions from the molecular, cellular, to the whole–body levels. The main molecular pathways affected by CR include mTOR, insulin signaling, AMPK, and sirtuins. Members of the family of nuclear receptors, the three peroxisome proliferator–activated receptors (PPARs), PPARα, PPARβ/δ, and PPARγ take part in the modulation of these pathways.
- caloric restriction
- nuclear receptors
- circadian rhythm
- metabolism
1. Insulin Signaling in Metabolism and Circadian Rhythm Regulation
2. mTOR Signaling and the Circadian Rhythm
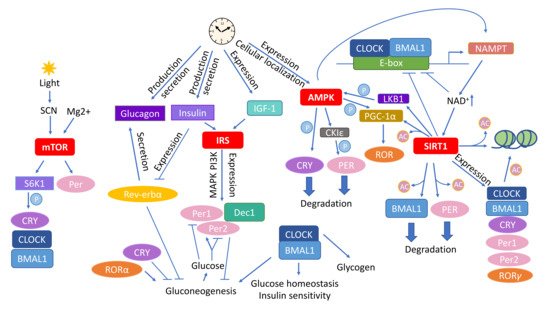
3. AMPK as an Energy Sensor and Executor of Circadian Metabolic Activities
4. SIRT Energy Sensors in the Context of CR and Daily Rhythmicity
This entry is adapted from the peer-reviewed paper 10.3390/nu12113476
References
- Bluher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574.
- Tatar, M.; Bartke, A.; Antebi, A. The endocrine regulation of aging by insulin-like signals. Science 2003, 299, 1346–1351.
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464.
- Taguchi, A.; Wartschow, L.M.; White, M.F. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science 2007, 317, 369–372.
- Selman, C.; Lingard, S.; Choudhury, A.I.; Batterham, R.L.; Claret, M.; Clements, M.; Ramadani, F.; Okkenhaug, K.; Schuster, E.; Blanc, E.; et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. FASEB J. 2008, 22, 807–818.
- Weindruch, R. The retardation of aging by caloric restriction: Studies in rodents and primates. Toxicol. Pathol. 1996, 24, 742–745.
- Kim, D.H.; Park, M.H.; Lee, E.K.; Choi, Y.J.; Chung, K.W.; Moon, K.M.; Kim, M.J.; An, H.J.; Park, J.W.; Kim, N.D.; et al. The roles of FoxOs in modulation of aging by calorie restriction. Biogerontology 2015, 16, 1–14.
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316.
- Rowland, A.F.; Fazakerley, D.J.; James, D.E. Mapping insulin/GLUT4 circuitry. Traffic 2011, 12, 672–681.
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10.
- Piscitello, D.; Varshney, D.; Lilla, S.; Vizioli, M.G.; Reid, C.; Gorbunova, V.; Seluanov, A.; Gillespie, D.A.; Adams, P.D. AKT overactivation can suppress DNA repair via p70S6 kinase-dependent downregulation of MRE11. Oncogene 2018, 37, 427–438.
- Jia, Y.; Song, W.; Zhang, F.; Yan, J.; Yang, Q. Akt1 inhibits homologous recombination in Brca1-deficient cells by blocking the Chk1-Rad51 pathway. Oncogene 2013, 32, 1943–1949.
- Liu, P.; Gan, W.; Guo, C.; Xie, A.; Gao, D.; Guo, J.; Zhang, J.; Willis, N.; Su, A.; Asara, J.M.; et al. Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Mol. Cell 2015, 57, 648–661.
- Astafev, A.A.; Patel, S.A.; Kondratov, R.V. Calorie restriction effects on circadian rhythms in gene expression are sex dependent. Sci. Rep. 2017, 7, 9716.
- Zhang, E.E.; Liu, A.C.; Hirota, T.; Miraglia, L.J.; Welch, G.; Pongsawakul, P.Y.; Liu, X.; Atwood, A.; Huss, J.W., 3rd; Janes, J.; et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell 2009, 139, 199–210.
- Patel, S.A.; Chaudhari, A.; Gupta, R.; Velingkaar, N.; Kondratov, R.V. Circadian clocks govern calorie restriction-mediated life span extension through BMAL1- and IGF-1-dependent mechanisms. FASEB J. 2016, 30, 1634–1642.
- Yamajuku, D.; Inagaki, T.; Haruma, T.; Okubo, S.; Kataoka, Y.; Kobayashi, S.; Ikegami, K.; Laurent, T.; Kojima, T.; Noutomi, K.; et al. Real-time monitoring in three-dimensional hepatocytes reveals that insulin acts as a synchronizer for liver clock. Sci. Rep. 2012, 2, 439.
- Tahara, Y.; Otsuka, M.; Fuse, Y.; Hirao, A.; Shibata, S. Refeeding after fasting elicits insulin-dependent regulation of Per2 and Rev-erbalpha with shifts in the liver clock. J. Biol. Rhythms 2011, 26, 230–240.
- Lamia, K.A.; Sachdeva, U.M.; DiTacchio, L.; Williams, E.C.; Alvarez, J.G.; Egan, D.F.; Vasquez, D.S.; Juguilon, H.; Panda, S.; Shaw, R.J.; et al. AMPK regulates the circadian clock by cryptochrome phosphorylation and degradation. Science 2009, 326, 437–440.
- Iwanaga, H.; Yano, M.; Miki, H.; Okada, K.; Azama, T.; Takiguchi, S.; Fujiwara, Y.; Yasuda, T.; Nakayama, M.; Kobayashi, M.; et al. Per2 gene expressions in the suprachiasmatic nucleus and liver differentially respond to nutrition factors in rats. JPEN J. Parenter. Enteral. Nutr. 2005, 29, 157–161.
- Challet, E.; Losee-Olson, S.; Turek, F.W. Reduced glucose availability attenuates circadian responses to light in mice. Am. J. Physiol. 1999, 276, R1063–R1070.
- Hirota, T.; Okano, T.; Kokame, K.; Shirotani-Ikejima, H.; Miyata, T.; Fukada, Y. Glucose down-regulates Per1 and Per2 mRNA levels and induces circadian gene expression in cultured Rat-1 fibroblasts. J. Biol. Chem. 2002, 277, 44244–44251.
- Marcheva, B.; Ramsey, K.M.; Buhr, E.D.; Kobayashi, Y.; Su, H.; Ko, C.H.; Ivanova, G.; Omura, C.; Mo, S.; Vitaterna, M.H.; et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010, 466, 627–631.
- Vieira, E.; Marroqui, L.; Figueroa, A.L.; Merino, B.; Fernandez-Ruiz, R.; Nadal, A.; Burris, T.P.; Gomis, R.; Quesada, I. Involvement of the clock gene Rev-erb alpha in the regulation of glucagon secretion in pancreatic alpha-cells. PLoS ONE 2013, 8, e69939.
- Sadacca, L.A.; Lamia, K.A.; deLemos, A.S.; Blum, B.; Weitz, C.J. An intrinsic circadian clock of the pancreas is required for normal insulin release and glucose homeostasis in mice. Diabetologia 2011, 54, 120–124.
- Vieira, E.; Merino, B.; Quesada, I. Role of the clock gene Rev-erbalpha in metabolism and in the endocrine pancreas. Diabetes Obes. Metab. 2015, 17 (Suppl. S1), 106–114.
- Muhlbauer, E.; Wolgast, S.; Finckh, U.; Peschke, D.; Peschke, E. Indication of circadian oscillations in the rat pancreas. FEBS Lett. 2004, 564, 91–96.
- Rudic, R.D.; McNamara, P.; Curtis, A.M.; Boston, R.C.; Panda, S.; Hogenesch, J.B.; Fitzgerald, G.A. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2004, 2, e377.
- Barclay, J.L.; Shostak, A.; Leliavski, A.; Tsang, A.H.; Johren, O.; Muller-Fielitz, H.; Landgraf, D.; Naujokat, N.; van der Horst, G.T.; Oster, H. High-fat diet-induced hyperinsulinemia and tissue-specific insulin resistance in Cry-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1053–E1063.
- Zhao, Y.; Zhang, Y.; Zhou, M.; Wang, S.; Hua, Z.; Zhang, J. Loss of mPer2 increases plasma insulin levels by enhanced glucose-stimulated insulin secretion and impaired insulin clearance in mice. FEBS Lett. 2012, 586, 1306–1311.
- Delezie, J.; Dumont, S.; Dardente, H.; Oudart, H.; Grechez-Cassiau, A.; Klosen, P.; Teboul, M.; Delaunay, F.; Pevet, P.; Challet, E. The nuclear receptor REV-ERBalpha is required for the daily balance of carbohydrate and lipid metabolism. FASEB J. 2012, 26, 3321–3335.
- Kalsbeek, A.; la Fleur, S.; Fliers, E. Circadian control of glucose metabolism. Mol. Metab. 2014, 3, 372–383.
- Doi, R.; Oishi, K.; Ishida, N. CLOCK regulates circadian rhythms of hepatic glycogen synthesis through transcriptional activation of Gys2. J. Biol. Chem. 2010, 285, 22114–22121.
- Matsuoka, H.; Shima, A.; Kuramoto, D.; Kikumoto, D.; Matsui, T.; Michihara, A. Phosphoenolpyruvate carboxykinase, a key enzyme that controls blood glucose, is a target of retinoic acid receptor-related orphan receptor alpha. PLoS ONE 2015, 10, e0137955.
- Kadiri, S.; Monnier, C.; Ganbold, M.; Ledent, T.; Capeau, J.; Antoine, B. The nuclear retinoid-related orphan receptor-alpha regulates adipose tissue glyceroneogenesis in addition to hepatic gluconeogenesis. Am. J. Physiol Endocrinol. Metab. 2015, 309, E105–E114.
- Kojetin, D.; Wang, Y.; Kamenecka, T.M.; Burris, T.P. Identification of SR8278, a synthetic antagonist of the nuclear heme receptor REV-ERB. ACS Chem. Biol. 2011, 6, 131–134.
- Yin, L.; Wu, N.; Curtin, J.C.; Qatanani, M.; Szwergold, N.R.; Reid, R.A.; Waitt, G.M.; Parks, D.J.; Pearce, K.H.; Wisely, G.B.; et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science 2007, 318, 1786–1789.
- Zhang, E.E.; Liu, Y.; Dentin, R.; Pongsawakul, P.Y.; Liu, A.C.; Hirota, T.; Nusinow, D.A.; Sun, X.; Landais, S.; Kodama, Y.; et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 2010, 16, 1152–1156.
- Lamia, K.A.; Papp, S.J.; Yu, R.T.; Barish, G.D.; Uhlenhaut, N.H.; Jonker, J.W.; Downes, M.; Evans, R.M. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature 2011, 480, 552–556.
- Zani, F.; Breasson, L.; Becattini, B.; Vukolic, A.; Montani, J.P.; Albrecht, U.; Provenzani, A.; Ripperger, J.A.; Solinas, G. PER2 promotes glucose storage to liver glycogen during feeding and acute fasting by inducing Gys2 PTG and G L expression. Mol. Metab. 2013, 2, 292–305.
- Bhaskar, P.T.; Hay, N. The two TORCs and Akt. Dev. Cell 2007, 12, 487–502.
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484.
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468.
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128.
- Corradetti, M.N.; Guan, K.L. Upstream of the mammalian target of rapamycin: Do all roads pass through mTOR? Oncogene 2006, 25, 6347–6360.
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945.
- Arsham, A.M.; Neufeld, T.P. Thinking globally and acting locally with TOR. Curr. Opin. Cell Biol. 2006, 18, 589–597.
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183.
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014.
- Kim, J.E.; Chen, J. regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756.
- Zhang, H.H.; Huang, J.; Duvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.L.; Manning, B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE 2009, 4, e6189.
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236.
- Wang, B.T.; Ducker, G.S.; Barczak, A.J.; Barbeau, R.; Erle, D.J.; Shokat, K.M. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc. Natl. Acad. Sci. USA 2011, 108, 15201–15206.
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71.
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797.
- Cao, R.; Lee, B.; Cho, H.Y.; Saklayen, S.; Obrietan, K. Photic regulation of the mTOR signaling pathway in the suprachiasmatic circadian clock. Mol. Cell Neurosci. 2008, 38, 312–324.
- Cao, R.; Anderson, F.E.; Jung, Y.J.; Dziema, H.; Obrietan, K. Circadian regulation of mammalian target of rapamycin signaling in the mouse suprachiasmatic nucleus. Neuroscience 2011, 181, 79–88.
- Cornu, M.; Oppliger, W.; Albert, V.; Robitaille, A.M.; Trapani, F.; Quagliata, L.; Fuhrer, T.; Sauer, U.; Terracciano, L.; Hall, M.N. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc. Natl. Acad. Sci. USA 2014, 111, 11592–11599.
- Jouffe, C.; Cretenet, G.; Symul, L.; Martin, E.; Atger, F.; Naef, F.; Gachon, F. The circadian clock coordinates ribosome biogenesis. PLoS Biol. 2013, 11, e1001455.
- Khapre, R.V.; Kondratova, A.A.; Patel, S.; Dubrovsky, Y.; Wrobel, M.; Antoch, M.P.; Kondratov, R.V. BMAL1-dependent regulation of the mTOR signaling pathway delays aging. Aging 2014, 6, 48–57.
- Lipton, J.O.; Yuan, E.D.; Boyle, L.M.; Ebrahimi-Fakhari, D.; Kwiatkowski, E.; Nathan, A.; Guttler, T.; Davis, F.; Asara, J.M.; Sahin, M. The circadian protein BMAL1 regulates translation in response to S6K1-mediated phosphorylation. Cell 2015, 161, 1138–1151.
- Huang, C.C.; Ko, M.L.; Ko, G.Y. A new functional role for mechanistic/mammalian target of rapamycin complex 1 (mTORC1) in the circadian regulation of L-type voltage-gated calcium channels in avian cone photoreceptors. PLoS ONE 2013, 8, e73315.
- Dragert, K.; Bhattacharya, I.; Hall, M.N.; Humar, R.; Battegay, E.; Haas, E. Basal mTORC2 activity and expression of its components display diurnal variation in mouse perivascular adipose tissue. Biochem. Biophys. Res. Commun. 2016, 473, 317–322.
- Chang, S.W.; Yoshihara, T.; Machida, S.; Naito, H. Circadian rhythm of intracellular protein synthesis signaling in rat cardiac and skeletal muscles. Biochem. Biophys. Rep. 2017, 9, 153–158.
- Saraf, A.; Luo, J.; Morris, D.R.; Storm, D.R. Phosphorylation of eukaryotic translation initiation factor 4E and eukaryotic translation initiation factor 4E-binding protein (4EBP) and their upstream signaling components undergo diurnal oscillation in the mouse hippocampus: Implications for memory persistence. J. Biol. Chem. 2014, 289, 20129–20138.
- Albert, V.; Cornu, M.; Hall, M.N. mTORC1 signaling in Agrp neurons mediates circadian expression of Agrp and NPY but is dispensable for regulation of feeding behavior. Biochem. Biophys. Res. Commun. 2015, 464, 480–486.
- Khapre, R.V.; Patel, S.A.; Kondratova, A.A.; Chaudhary, A.; Velingkaar, N.; Antoch, M.P.; Kondratov, R.V. Metabolic clock generates nutrient anticipation rhythms in mTOR signaling. Aging 2014, 6, 675–689.
- Zheng, X.; Sehgal, A. AKT and TOR signaling set the pace of the circadian pacemaker. Curr. Biol. 2010, 20, 1203–1208.
- Kijak, E.; Pyza, E. TOR signaling pathway and autophagy are involved in the regulation of circadian rhythms in behavior and plasticity of L2 interneurons in the brain of Drosophila melanogaster. PLoS ONE 2017, 12, e0171848.
- Cao, R.; Robinson, B.; Xu, H.; Gkogkas, C.; Khoutorsky, A.; Alain, T.; Yanagiya, A.; Nevarko, T.; Liu, A.C.; Amir, S.; et al. Translational control of entrainment and synchrony of the suprachiasmatic circadian clock by mTOR/4E-BP1 signaling. Neuron 2013, 79, 712–724.
- Cao, R.; Li, A.; Cho, H.Y.; Lee, B.; Obrietan, K. Mammalian target of rapamycin signaling modulates photic entrainment of the suprachiasmatic circadian clock. J. Neurosci. 2010, 30, 6302–6314.
- Ramanathan, C.; Kathale, N.D.; Liu, D.; Lee, C.; Freeman, D.A.; Hogenesch, J.B.; Cao, R.; Liu, A.C. mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 2018, 14, e1007369.
- Feeney, K.A.; Hansen, L.L.; Putker, M.; Olivares-Yanez, C.; Day, J.; Eades, L.J.; Larrondo, L.F.; Hoyle, N.P.; O’Neill, J.S.; van Ooijen, G. Daily magnesium fluxes regulate cellular timekeeping and energy balance. Nature 2016, 532, 375–379.
- Hatori, M.; Vollmers, C.; Zarrinpar, A.; DiTacchio, L.; Bushong, E.A.; Gill, S.; Leblanc, M.; Chaix, A.; Joens, M.; Fitzpatrick, J.A.; et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab. 2012, 15, 848–860.
- Winder, W.W.; Hardie, D.G. AMP-activated protein kinase, a metabolic master switch: Possible roles in type 2 diabetes. Am. J. Physiol. 1999, 277, E1–E10.
- Koo, S.H.; Flechner, L.; Qi, L.; Zhang, X.; Screaton, R.A.; Jeffries, S.; Hedrick, S.; Xu, W.; Boussouar, F.; Brindle, P.; et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature 2005, 437, 1109–1111.
- Chen, S.; Murphy, J.; Toth, R.; Campbell, D.G.; Morrice, N.A.; Mackintosh, C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem. J. 2008, 409, 449–459.
- Geraghty, K.M.; Chen, S.; Harthill, J.E.; Ibrahim, A.F.; Toth, R.; Morrice, N.A.; Vandermoere, F.; Moorhead, G.B.; Hardie, D.G.; MacKintosh, C. Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem. J. 2007, 407, 231–241.
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022.
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008, 57, 860–867.
- Davies, S.P.; Sim, A.T.; Hardie, D.G. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 1990, 187, 183–190.
- Davies, S.P.; Carling, D.; Munday, M.R.; Hardie, D.G. Diurnal rhythm of phosphorylation of rat liver acetyl-CoA carboxylase by the AMP-activated protein kinase, demonstrated using freeze-clamping. Effects of high fat diets. Eur. J. Biochem. 1992, 203, 615–623.
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388.
- Muoio, D.M.; Seefeld, K.; Witters, L.A.; Coleman, R.A. AMP-activated kinase reciprocally regulates triacylglycerol synthesis and fatty acid oxidation in liver and muscle: Evidence that sn-glycerol-3-phosphate acyltransferase is a novel target. Biochem. J. 1999, 338, 783–791.
- Clarke, P.R.; Hardie, D.G. Regulation of HMG-CoA reductase: Identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990, 9, 2439–2446.
- Hoppe, S.; Bierhoff, H.; Cado, I.; Weber, A.; Tiebe, M.; Grummt, I.; Voit, R. AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc. Natl. Acad. Sci. USA 2009, 106, 17781–17786.
- Marsin, A.S.; Bouzin, C.; Bertrand, L.; Hue, L. The stimulation of glycolysis by hypoxia in activated monocytes is mediated by AMP-activated protein kinase and inducible 6-phosphofructo-2-kinase. J. Biol. Chem. 2002, 277, 30778–30783.
- Jorgensen, S.B.; Nielsen, J.N.; Birk, J.B.; Olsen, G.S.; Viollet, B.; Andreelli, F.; Schjerling, P.; Vaulont, S.; Hardie, D.G.; Hansen, B.F.; et al. The alpha2-5’AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes 2004, 53, 3074–3081.
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262.
- Leclerc, I.; Lenzner, C.; Gourdon, L.; Vaulont, S.; Kahn, A.; Viollet, B. Hepatocyte nuclear factor-4alpha involved in type 1 maturity-onset diabetes of the young is a novel target of AMP-activated protein kinase. Diabetes 2001, 50, 1515–1521.
- Lee, J.M.; Seo, W.Y.; Song, K.H.; Chanda, D.; Kim, Y.D.; Kim, D.K.; Lee, M.W.; Ryu, D.; Kim, Y.H.; Noh, J.R.; et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB.CRTC2 complex by orphan nuclear receptor small heterodimer partner. J. Biol. Chem. 2010, 285, 32182–32191.
- Dufer, M.; Noack, K.; Krippeit-Drews, P.; Drews, G. Activation of the AMP-activated protein kinase enhances glucose-stimulated insulin secretion in mouse beta-cells. Islets 2010, 2, 156–163.
- Chavez, J.A.; Roach, W.G.; Keller, S.R.; Lane, W.S.; Lienhard, G.E. Inhibition of GLUT4 translocation by Tbc1d1, a Rab GTPase-activating protein abundant in skeletal muscle, is partially relieved by AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 9187–9195.
- Steinberg, G.R.; Kemp, B.E. AMPK in health and disease. Physiol. Rev. 2009, 89, 1025–1078.
- Suzuki, A.; Okamoto, S.; Lee, S.; Saito, K.; Shiuchi, T.; Minokoshi, Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol. Cell Biol. 2007, 27, 4317–4327.
- Um, J.H.; Pendergast, J.S.; Springer, D.A.; Foretz, M.; Viollet, B.; Brown, A.; Kim, M.K.; Yamazaki, S.; Chung, J.H. AMPK regulates circadian rhythms in a tissue- and isoform-specific manner. PLoS ONE 2011, 6, e18450.
- Vieira, E.; Nilsson, E.C.; Nerstedt, A.; Ormestad, M.; Long, Y.C.; Garcia-Roves, P.M.; Zierath, J.R.; Mahlapuu, M. Relationship between AMPK and the transcriptional balance of clock-related genes in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1032–E1037.
- Um, J.H.; Yang, S.; Yamazaki, S.; Kang, H.; Viollet, B.; Foretz, M.; Chung, J.H. Activation of 5’-AMP-activated kinase with diabetes drug metformin induces casein kinase Iepsilon (CKIepsilon)-dependent degradation of clock protein mPer2. J. Biol. Chem. 2007, 282, 20794–20798.
- Grimaldi, B.; Sassone-Corsi, P. Circadian rhythms: Metabolic clockwork. Nature 2007, 447, 386–387.
- Liu, C.; Li, S.; Liu, T.; Borjigin, J.; Lin, J.D. Transcriptional coactivator PGC-1alpha integrates the mammalian clock and energy metabolism. Nature 2007, 447, 477–481.
- Minokoshi, Y.; Alquier, T.; Furukawa, N.; Kim, Y.B.; Lee, A.; Xue, B.; Mu, J.; Foufelle, F.; Ferre, P.; Birnbaum, M.J.; et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 2004, 428, 569–574.
- Andersson, U.; Filipsson, K.; Abbott, C.R.; Woods, A.; Smith, K.; Bloom, S.R.; Carling, D.; Small, C.J. AMP-activated protein kinase plays a role in the control of food intake. J. Biol. Chem. 2004, 279, 12005–12008.
- Liou, G.G.; Tanny, J.C.; Kruger, R.G.; Walz, T.; Moazed, D. Assembly of the SIR complex and its regulation by O-acetyl-ADP-ribose, a product of NAD-dependent histone deacetylation. Cell 2005, 121, 515–527.
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 2004, 16, 93–105.
- Dang, W. The controversial world of sirtuins. Drug. Discov. Today Technol. 2014, 12, e9–e17.
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238.
- Shimazu, T.; Hirschey, M.D.; Hua, L.; Dittenhafer-Reed, K.E.; Schwer, B.; Lombard, D.B.; Li, Y.; Bunkenborg, J.; Alt, F.W.; Denu, J.M.; et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010, 12, 654–661.
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125.
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667.
- Preyat, N.; Leo, O. Sirtuin deacylases: A molecular link between metabolism and immunity. J. Leukoc. Biol. 2013, 93, 669–680.
- Satoh, A.; Brace, C.S.; Ben-Josef, G.; West, T.; Wozniak, D.F.; Holtzman, D.M.; Herzog, E.D.; Imai, S. SIRT1 promotes the central adaptive response to diet restriction through activation of the dorsomedial and lateral nuclei of the hypothalamus. J. Neurosci. 2010, 30, 10220–10232.
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580.
- Whitaker, R.; Faulkner, S.; Miyokawa, R.; Burhenn, L.; Henriksen, M.; Wood, J.G.; Helfand, S.L. Increased expression of Drosophila Sir2 extends life span in a dose-dependent manner. Aging 2013, 5, 682–691.
- Lin, S.J.; Defossez, P.A.; Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000, 289, 2126–2128.
- Rogina, B.; Helfand, S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl. Acad. Sci. USA 2004, 101, 15998–16003.
- Tissenbaum, H.A.; Guarente, L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature 2001, 410, 227–230.
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 2008, 135, 907–918.
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118.
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1. J. Biol. Chem. 2005, 280, 16456–16460.
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell 2006, 127, 1109–1122.
- Daitoku, H.; Hatta, M.; Matsuzaki, H.; Aratani, S.; Ohshima, T.; Miyagishi, M.; Nakajima, T.; Fukamizu, A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10042–10047.
- van der Horst, A.; Tertoolen, L.G.; de Vries-Smits, L.M.; Frye, R.A.; Medema, R.H.; Burgering, B.M. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1). J. Biol. Chem. 2004, 279, 28873–28879.
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015.
- Frescas, D.; Valenti, L.; Accili, D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J. Biol. Chem. 2005, 280, 20589–20595.
- Accili, D.; Arden, K.C. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 2004, 117, 421–426.
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207.
- Gross, D.N.; van den Heuvel, A.P.; Birnbaum, M.J. The role of FoxO in the regulation of metabolism. Oncogene 2008, 27, 2320–2336.
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060.
- Fulco, M.; Cen, Y.; Zhao, P.; Hoffman, E.P.; McBurney, M.W.; Sauve, A.A.; Sartorelli, V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 2008, 14, 661–673.
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026.
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635.
- Asher, G.; Gatfield, D.; Stratmann, M.; Reinke, H.; Dibner, C.; Kreppel, F.; Mostoslavsky, R.; Alt, F.W.; Schibler, U. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 2008, 134, 317–328.
- Rutter, J.; Reick, M.; Wu, L.C.; McKnight, S.L. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science 2001, 293, 510–514.
- Oike, H.; Kobori, M. Resveratrol regulates circadian clock genes in Rat-1 fibroblast cells. Biosci. Biotechnol. Biochem. 2008, 72, 3038–3040.
- Sun, L.; Wang, Y.; Song, Y.; Cheng, X.R.; Xia, S.; Rahman, M.R.; Shi, Y.; Le, G. Resveratrol restores the circadian rhythmic disorder of lipid metabolism induced by high-fat diet in mice. Biochem. Biophys. Res. Commun. 2015, 458, 86–91.
- Miranda, J.; Portillo, M.P.; Madrid, J.A.; Arias, N.; Macarulla, M.T.; Garaulet, M. Effects of resveratrol on changes induced by high-fat feeding on clock genes in rats. Br. J. Nutr. 2013, 110, 1421–1428.
- Nakahata, Y.; Kaluzova, M.; Grimaldi, B.; Sahar, S.; Hirayama, J.; Chen, D.; Guarente, L.P.; Sassone-Corsi, P. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell 2008, 134, 329–340.
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654.
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657.
- Peek, C.B.; Affinati, A.H.; Ramsey, K.M.; Kuo, H.Y.; Yu, W.; Sena, L.A.; Ilkayeva, O.; Marcheva, B.; Kobayashi, Y.; Omura, C.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013, 342, 1243417.
- Sato, S.; Solanas, G.; Peixoto, F.O.; Bee, L.; Symeonidi, A.; Schmidt, M.S.; Brenner, C.; Masri, S.; Benitah, S.A.; Sassone-Corsi, P. Circadian reprogramming in the liver identifies metabolic pathways of aging. Cell 2017, 170, 664–677.e611.
- Sahar, S.; Masubuchi, S.; Eckel-Mahan, K.; Vollmer, S.; Galla, L.; Ceglia, N.; Masri, S.; Barth, T.K.; Grimaldi, B.; Oluyemi, O.; et al. Circadian control of fatty acid elongation by SIRT1 protein-mediated deacetylation of acetyl-coenzyme A synthetase 1. J. Biol. Chem. 2014, 289, 6091–6097.
- Hallows, W.C.; Lee, S.; Denu, J.M. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc. Natl. Acad. Sci. USA 2006, 103, 10230–10235.