Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Still’s disease and Kawasaki disease (KD) today belong to the group of cytokine storm syndromes, a pathophysiological set related to excessive activation of the innate immune response.
- Kawasaki disease
- Still’s disease
- disease overlap
- interleukin-1
1. Conceptual Evolution of Still’s Disease
Sir Frederick Still described at the end of the 19th century (1897), a primary rheumatic condition remarkably different from rheumatoid arthritis with “a chronic progressive enlargement of joints, associated with general enlargement of glands and enlargement of spleen” and with the onset “in young children almost always before the second dentition.” He also noted in his report the presence of fever, chills, anaemia and stunted growth in a subset of patients [1]. It was not until 1946, with the advent of paediatric rheumatology by pioneers such as Mayer S. Diamantberger and Barbara Ansell, that the heterogeneity of juvenile arthritis was described, in which Still’s disease appeared as a separate subgroup [2,3]. The major distinguishing features of Still’s disease, in addition to those previously described, were the severity of joint involvement in 50% of cases with predominant involvement of the hips, knees and wrists. It was during the 1980s that a first alert on cases of severe hepatic and neurological complications, occurring after an infection or the use of certain drugs in children, made it possible to make the association between Still’s disease and macrophage activation syndrome (MAS), a peculiar entity neighbour of inherited hemophagocytic lymphohistiocytosis (HLH) [4]. Over time, a very specific treatment response profile was also identified, with frequent dependence on corticosteroids, little benefit from treatment with methotrexate and later with anti TNF, the first biotherapies available for JIA in the early 1990s [5]. This resistance to treatments echoed the work of Dayer and de Benedetti who had pointed out years earlier the predominant role of interleukin 6 (IL-6) in the disease pathogenesis [6,7]. Other studies then highlighted the role of interleukin 1 (IL-1) in systemic manifestations, and first attempt of IL-1 blockade by anakinra have triggered hopes and expectations [8].
In parallel with all this, the concept of auto inflammation emerged in 1999 with the discovery of the TNF receptor 1 (TNFR1) deficiency syndrome, an inherited hyper inflammatory condition, linked to aberrant and deregulated IL-1 secretion induced by intracellular stress related to a defect in folding and clearance of the mutated TNFR1 [9]. Thus, other hereditary and multifactorial diseases, presenting with similar inflammatory characteristics, mediated by IL-1, gradually entered this category. Briefly, patients experience acute or chronic inflammatory symptoms including fever, malaise, myalgia, skin rashes, and abdominal pain.
2. Conceptual Evolution of Kawasaki Disease
In 1961 in Japan, in a 4-year-old boy with a 2-week fever, Dr Tomisaku Kawasaki saw for the first time non-purulent conjunctivitis, red, dry, and fissured lips, a raspberry tongue, cervical lymphadenopathy, erythema multiforme, and changes in the extremities (redness, swelling of the palms and soles). It was not until another 50 cases were described that a new disease was recognized and named after him [11]. The associated coronary artery aneurysms (CAA) were not identified until 1970. Initially, Kawasaki disease (KD) appeared to be an infectious disease, but to date, no single causative agent has been identified. Before intravenous immunoglobulins (IVIG) treatment became the gold standard, the incidence of CAA was 25% [12].
KD is a systemic inflammatory disease affecting primarily the medium-sized vessels and occurring in more than 75% of cases before the age of 5 years. The disease generally follows or is concomitant of upper respiratory or gastrointestinal symptoms, and manifests as persistent spiking fevers; associated with muco cutaneous symptoms (generalized skin rash, eye redness, strawberry tongue, oral cavity erythema, redness and edema at the hands and feet, and enlarged cervical lymph nodes) [13]. At onset, a biological cytokine storm containing, IL-1, IL-6, TNF, IL-17A, chemokines and metalloproteases, is sometimes present going as far as MAS, and its intensity goes hand in hand with the development of CAA, the most feared complication [14]. Studies carried out on the murine model of KD vasculitis have led to a better understanding of the role of IL-1 and the NLRP3 inflammasome in the induction of endothelial cells injury causing aneurysms [15]. The role of IL-1 in human KD is now largely supported and the IL-1 blockade could be one of the mechanisms of action of IVIG, the standard treatment of KD. In addition, exploratory trials are developing for better defining the full effects and the place of IL-1 blocking agents in KD treatment strategy [16].
3. Still’s Disease and KD, How Much Are They Related?
Although it is not clear what triggers them, it appears that Still’s disease and KD occur in the same age groups with a maximum of cases around the age of 2 to 4 years and rarer cases at adolescence and in young adults [17,18,19,20]. Both correspond to a deregulation of the innate immune response towards an excessive inflammation following a still unknown stimulation, coming from the airways or the digestive system. The involvement of the oropharyngeal sphere is probably a critical point that could link Still ‘s disease and KD to another multifactorial autoinflammatory syndrome, namely PFAPA syndrome, which also occurs in the same age groups [21]. Other clinical symptoms, spiking fever, rash, hepatomegaly, adenopathy, and cardiac involvement are very similar between KD and Still’s diseas.
In addition, it is not uncommon in the paediatric experience for patients to develop Still’s disease after an initial presentation of KD including coronary dilations [25]. Patients in both diseases develop severe biological inflammation with hyperleukocytosis, thrombocytosis, anaemia, hypoalbuminemia, and hepatic cytolysis. Other biomarkers of inflammation such as, ferritin, IL-1β, IL-6, TNF, IFNγ and S100 proteins are elevated and correlate with disease activity in both diseases and cardiac complications in KD [26,27]. MAS is almost a hallmark of Still’s disease in which, it is present subclinically in 40% of cases and apparent in 7% [17]. MAS may also occur during the acute phase of KD with an estimate of 1% of cases; the actual incidence is still unknown because it is more often subclinical and exceptionally apparent and life threatening [28].
4. How Their Pathophysiology Could Be Similar?
Both diseases involve a cytokine storm, a type of aberrant innate immune response as described in systemic forms of COVID-19 (multisystem inflammatory syndrome) and shared by other as yet poorly understood conditions such as SAM, HLH, cytokine release syndrome, toxic shock syndrome and acute respiratory distress syndrome [32]. Infectious organisms are capable of triggering onset of both diseases in genetically predisposed children, affected mainly between 6 months and 5 years of age. The peak age of diseases onset coincides with the period during which children are most susceptible to common pathogens. Microbial danger signals (PAMPs) recognize innate immune receptors (TLRs, NOD2, NLRP3) that enhance numerous danger signals (DAMPs: S100 proteins, metalloproteases, pro IL-1β) and secondary activation of canonic pathways cytokines secretion, in which both the NFKB and inflammasome pathways are involved (Figure 1) [33]. Among genetic susceptibility factors of KD, single-nucleotide polymorphisms in ITPKC encoding for an inositol-trisphosphate 3-kinase, regulating negatively the opening of calcium channels, induce an increase of calcium release with two downstream consequences; 1/the activation of the calpain pathway enhancing the interleukin 1α a potent danger signal and 2/the activation of the NLRP3 inflammasome secondary to increased secretion of ROS (reactive oxygen species) [34]. Susceptibility to Still’s disease and response to IL-1 blockage treatment have recently been linked with IL1-receptor antagonist gene (IL1RN) polymorphisms [35]. MAS is a common complication in both diseases and is generally linked with either: onset of disease, disease activity or coinfection as a second hit.
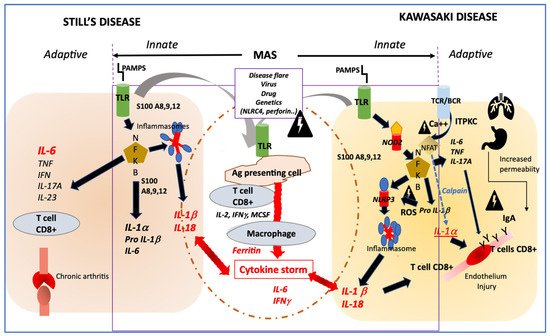
Figure 1. Profiles of immune activation are similar in Still’s disease and Kawasaki disease. Innate immune system: In both diseases danger signals coming from outside (PAMPS) sensed by TLR or from inside (DAMPS) coming from activated macrophages (such as S100 proteins) may activate both the NFKB and the NLRP3 inflammasome pathways that drives the secretion of IL-1β and IL-18. In both diseases, additional triggering event such as disease flare, infection, in the context of genetic modifiers, may induce an amplification cascade of inflammation resulting in MAS. Adaptive immune system: In KD, the adaptive immune system may be activated primarily through antigen recognition by the BCR and TCR, and genetic variation in ITPKC may impair the regulation of calcium fluxes and then remove the inhibition of NFAT, which induces the secretion of IL-1α, a potent inducer of endothelial cells activation. In both Still’s disease and KD, NFKB activation induces an increased secretion of IL-6 and IL-17A, which participates in one case in bone erosion and inflammation and in the other case in endothelial inflammation and damage. Involvement of IgA from increased intestinal permeability and/or respiratory tract may participate to endothelial injury in KD. PAMPS: pathogen associated molecular patterns; DAMPS; danger associated molecular pattern; TLR; Toll-like receptor; NFKB; nuclear factor; NLRP3: NOD-like receptor family, pyrin domain containing 3; MAS: macrophage activation syndrome; BCR: B cells receptor; TCR: T cells receptor; ITPKC: inositol-trisphosphate 3-kinase; NFAT: Nuclear factor of activated T-cells; ROS: reactive oxygen species; NOD2: nucleotide-binding oligomerization domain 2.
This entry is adapted from the peer-reviewed paper 10.3390/jcm10153244
This entry is offline, you can click here to edit this entry!