Nucleocytoviricota is a large group of double-stranded DNA viruses that fully or partially replicate in the host cytoplasm. Despite marked differences, including virion shape, genome length, and host range, viruses belonging to this phylum have some very conserved characteristics, including the gene expression profile. A temporal pattern of gene expression, also known as a transcription cascade model, is described for these viruses, and comparative transcriptome analysis can be used as a starting point for future transcriptomic investigations.
- NCLDV
- transcription
- giant viruses
- gene expression
- evolution
1. Introduction
The nucleocytoplasmic large DNA viruses (NCLDV) are characterized by sharing a set of conserved genes related to replication, transcription, and morphogenesis, a phenomenon that suggests these viruses have a common evolutionary origin. Subsequent analyses of sequenced genomes of isolated viruses belonging to new families have supported the monophyly of the NCLDV group [1]. Initial comparative genomic analyses have derived a set of 40 core genes common to NCLDV. Among these core genes, there are only a few conserved in all known viruses that belong to this group, including the DNA polymerase B family, helicase-primase, and the late transcription factor 3 [2][3][4]. This set of genes called “hallmark genes”, reconstructed as present in the common ancestor, seem to have origins from different sources, the majority being homologous to eukaryotic genes and a small part derived from bacteriophage genes [3].
Distinct viral families are part of the NCLDV group, including Poxviridae, Asfarviridae, Iridoviridae, Phycodnaviridae, Ascoviridae, Mimiviridae, and Marseilleviridae. NCLDV have recently been officially classified into a new phylum, namely Nucleocytoviricota [5]. Other recently discovered giant viruses such as pandoravirus, faustovirus, kaumoebavirus, cedratvirus, pithovirus, mollivirus, pacmanvirus, orpheovirus, and medusavirus have also been included as members of the NCLDV group, despite not being officially classified into any existent taxa [6][7][8][9][10][11][12][13][14]. This group has unique characteristics such as large particles and genomes, which encode proteins that have never been described in other viruses [15]. Another characteristic shared by NCLDV is the fact that they replicate entirely or partially in the host cell’s cytoplasm, in which some viral groups exhibiting little dependence on the host cell’s transcriptional machinery, such as poxviruses and mimiviruses [15][16]. The presence of a robust transcriptional apparatus in some NCLDV has raised discussion about the origin and evolution of these viruses and their genomes [16]. In addition, their gene transcription is temporally regulated, allowing sets of genes to be classified as early, intermediate, or late in accordance with the stage of infection when they are transcribed. A previous study has divided the genes of NCLDV into clusters of orthologous groups (NCVOG), many of which could be assigned to putative functional classes [1].
2. Temporal Regulation of Gene Expression
Transcriptional regulation involves a sequence of steps and although most of them have been studied extensively using static biochemistry, much about the real-time kinetics of transcription has not been completely elucidated. As previously described, NCLDV have many characteristics in common. Among these characteristics, the transcription of their genes has a temporal profile, being classified as early, intermediate, and late; the genes expressed at each time point have different functions. The expression of each class of gene occurs in a cascade. This is due to the fact that the required transcription factors for the expression of each class of genes are the product of the genes previously expressed [17]. In that way, the products of some genes expressed early during the replication cycle of the virus will be required as transcription factors to induce the expression of other genes that will be expressed during the intermediate and later course of infection. Despite having a similar transcriptional regulation profile, the expression of the genes of each temporal class occurs at different times among NCLDV, due to the fact that their replication cycles last for different lengths of time. Nevertheless, the expression of genes from different time classes occurs at similar phases throughout the NCLDV multiplication cycle, that is, early, intermediate, and late during the course of the infection [16][17].
Such temporal classification has been possible due to the development of new techniques to quantify the expression levels of a large number of genes [18]. DNA microarrays were one of the first tools that allowed the large-scale study of the transcriptome. The technique is based on the hybridization of target strands on the complementary probe strand, allowing the identification of genes that are expressed at different times during the viral infection. A major limitation of this technique is to evaluate genes with low expression. Nevertheless, the advance of large-scale sequencing tools has allowed a more robust evaluation of the transcriptome of viruses. RNA sequencing (RNA-seq) has been widely used to study the gene expression profile of organisms and viruses, allowing a more in-depth comprehension of their transcriptional pattern. This technique provides complete sequencing of all expressed genes during the replication cycle of a virus, even if the genes have a low level of expression (depending on the coverage of the sequencing), allowing a better characterization of the virus’s transcriptome. A full description of the fundaments and possible usages of these techniques is beyond the scope of this review, but this topic has been extensively addressed elsewhere [19][20]. Although these tools are based on different technologies, both techniques make it possible to quantify the levels of gene expression and have had similar coverage in studies that have compared both techniques. Hence, they can be used in a comparison or as complementary methods in studies involving gene expression [18]. These techniques have been used to study the expression profile of different large and giant viruses, as will become apparent in the subsequent sections, providing important information about the biology of these viruses.
3. Transcription in Nucleocytoviricota
Among the viral families that comprise NCLDV, Poxviridae is by far the most studied group. Viruses belonging to this family have enveloped, ovoid-shaped particles that are 200 nm in diameter and 300 nm in length. Its genome consists of linear double-stranded DNA (dsDNA) of approximately 200 kilobase pairs (kbp), encoding 200 open reading frames (ORFs) [21]. This viral family comprises many human and other animal pathogens, including the one responsible for the most devastating disease that has affected humankind, the variola virus. The complexity of the poxvirus genome instigated speculation that these viruses synthesize their genome independently of the host nucleus. The transcription of poxvirus genes follows a temporal profile and is regulated by promoter regions and transcriptional factors. Thus, the genes are classified into early, intermediate, and late, and they are activated in a cascade sequence [16][17][22]. This temporal profile of gene transcription seems to be a common characteristic not only among members of the family, but also of other viruses related phylogenetically to poxviruses, such as asfarviruses (ASFV) that infect mammals.
Transcriptomic analysis of genes to classify them temporally, based on RNA-seq and CAGE-seq, identified 101 genes that show differences in their expression in early and late times of infection by ASFV. Only two-time points were evaluated, at 5 and 16 h post-infection (hpi); therefore, the genes were just classified as early or late, according to the evaluated moment of infection. Based on both techniques, 36 genes were classified as early—related to transcription, evasion of the host’s immune response, and DNA replication—and 55 as late—related to transcription, viral structure, morphogenesis, and DNA replication [23]. New transcriptomic analysis of ASFV, considering the entire time range of infection, would bring valuable information about the temporal classification of all genes, establishing a landscape for the transcriptome of this important swine pathogen.
Similar to other members of the phylum Nucleocytoviricota, iridoviruses (family Iridorividae) replication occurs partially in the host cell’s cytoplasm. Even with homologous RNA polymerase subunits in the iridoviruses genome, host cell RNA polymerase is necessary to synthesize early gene transcripts of viruses belonging to the Ranavirus and Iridovirus genera [16][24]. Thus, early mRNAs are synthesized in the nucleus using the host cell’s RNA polymerase II in the early stages. However, it is believed that the late transcripts are synthesized in the cytoplasm of the host cell by the RNA polymerase encoded by the virus [25]. Its gene expression is temporally regulated and results in the expression of three classes of genes: immediate-early, early, and late [25][26].
Unlike other NCLDV members, phycodnaviruses (family Phycodnaviridae) do not encode their own RNA polymerase: they depend on the host cell’s RNA polymerase for the transcription of their genes. Thus, viral DNA and associated proteins migrate to the nucleus to start transcription [27]. However, it has been reported that Emiliania huxleyi virus 86 (EhV-86) has six RNA polymerase subunits, suggesting that this particular member of the Phycodnaviridae family encodes its transcriptional apparatus and might have had a different evolutionary history compared with other relatives [28]. Transcriptomic data on Emiliania huxleyi virus 201 (EhV-201) showed that all six RNA polymerase subunits are expressed at distinct levels, but all at an early or early-late stage of the virus life cycle, possibly being key regulators of the transcription of late genes [29].
In 2003, the discovery of the first giant amoeba virus, Acanthamoeba polyphaga mimivirus (APMV), led to the establishment of the Mimiviridae family, also belonging to the NCLDV group [30]. Viruses belonging to this family have characteristics never before described in the virosphere, such as particles ~700 nm in diameter, capable of being visualized by optical microscopy. Their genome is composed of a dsDNA molecule of approximately 1.2 megabase pairs (Mbp), capable of encoding more than 1000 predicted proteins, and a wide range of elements related to transcription [31][32]. As with other NCLDV, mimiviruses also have a temporal transcription profile in which the transcribed genes are classified as early, intermediate, and late [33]. After the discovery of the first giant viruses and the increasing interest of the scientific community in this group, a great diversity of other giant viruses have been isolated, including Marseillevirus, faustovirus, pandoravirus, pithovirus, and mollivirus, among others. Studies aiming to elucidate how gene expression is regulated in most amoeba viruses are still needed.
4. Functional Comparative Analysis of Temporally Expressed Genes from Pathogens of Nucleocytoviricota
Marseillevirus T19 has a total of 457 genes[34]. 83 coding sequences (CDS) are classified as early genes (18%), 218 (48%) as intermediate, and 156 (34%) as late (Figure 1 A). The main functions observed for early genes of Marseillevirus are related to DNA replication and recombination, transcription and signal transduction, and some metabolic functions. Of the 83 genes classified as early, 3 (3.6%) are related to DNA replication and recombination. Most genes related to DNA replication are expressed at intermediate or late times during infection; however, some can be expressed as soon as the particle is internalized by the host cell. In addition, 7 (8.4%) genes are related to the transcription process, such as those encoding helicase and RNA polymerase subunits, 13 (15.6%) with regulation and signal transduction similar to some kinases, and only 1 (1%) is related to nucleotide metabolism (Figure 2). The transcription of early genes related to signal regulation and transduction, which encode serine/threonine kinase, suggests that the virus has the potential to manipulate its host’s responses, facilitating the establishment of productive infections. Regarding genes classified as intermediate, the main functions observed are also related to DNA replication and recombination in (25/218, 11.5%), followed by signal transduction regulation (8/218, 3.7%). Interestingly, many genes involved in DNA metabolism are also expressed during the late stages of the replication cycle, suggesting a continuous process of DNA manipulation during the virus life cycle. All of the genes related to virion structure and morphogenesis are expressed late, an expected feature considering that these genes are involved in the formation of new viral particles (Figure 2).
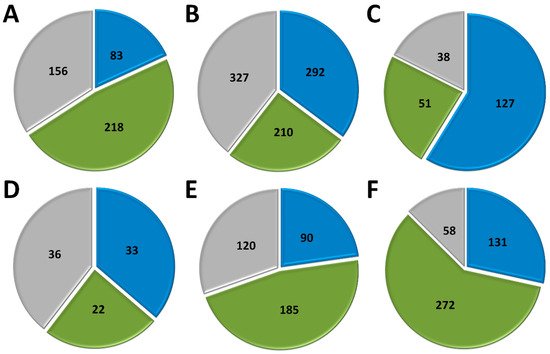
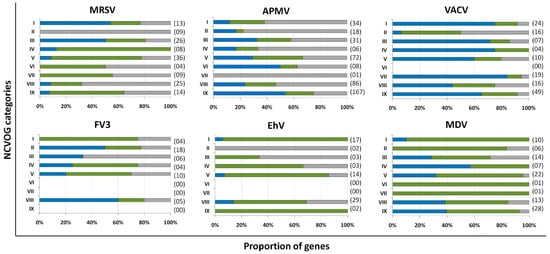
APMV has a total of 979 CDS, but for this analysis, we included only genes for which transcriptome data were available, for a total of 829 genes [33]. Approximately a third of APMV genes (292, 35.2%) are expressed early, 210 (25.3%) are intermediate and 327 (39.5%) are expressed in the final moments of the virus life cycle (Figure 1 B). Most of the genes related to DNA replication, recombination, and repair are classified as intermediate genes, but there are others expressed at other moments in the replication cycle, including DNA primase and some helicases (Figure 2). Genes involved in transcription and RNA processing, including transcription factors, are expressed early, while RNA polymerase subunits are predominantly expressed at intermediate and late times, possibly being important for further viral transcript synthesis. APMV encodes at least 31 genes involved in signal transduction regulation, including F-box domain-containing proteins and serine/threonine kinases, which are evenly distributed among distinct temporal classes of expression. A distinct feature of the APMV is the presence of translation-related genes, including aminoacyl-tRNA synthetases (aaRS). A total of eight CDS are members of the translational apparatus of APMV, with four being early expressed, including three aaRS, one intermediate (the remaining aaRS fall into this category, i.e., cysteinyl-tRNA synthetase), and three late genes (Figure 2). Similar to MRSV, most genes involved in the mimivirion structure are classified as late genes (14/18, 77.7%) (Figure 2). It is interesting to note that most of these genes are putative membrane genes, possibly involved in the formation of a virus factory and for establishing the initial steps of virion morphogenesis. The gene coding for the major capsid protein (L425) is a late gene.
Different from MRSV and APMV, VACV infects mammals and has been one of the most studied viruses throughout history [21][22]. Among the 218 genes, 118 (54.1%) are expressed at the initial moments of the replication cycle, while 51 (23.4%) and 38 (17.4%) are intermediate and late genes, respectively (Figure 1 C). It is interesting to note that VACV has many known genes related to the host-virus interaction, most of which (16/19, 84.2%) are expressed during the early moments of the virus life cycle (Figure 2). Among these genes are those related to the host immune response interaction, such as soluble interferon-alpha/beta receptor and chemokine-binding proteins. Most of the genes involved in the transcription and RNA processing of VACV are expressed early (18/24), while only four and two genes are expressed at intermediate and late phases of the replicative cycle, respectively (Figure 2). Genes involved in DNA replication, recombination, and repair are mostly expressed at the early and intermediate phases, and most genes related to virion morphogenesis are classified as intermediate and late genes (Figure 2). Although the functions of many VACV genes have been predicted, almost 40% of the genome remains uncharacterized and many genes have yet to be functionally evaluated, including several genes that have been annotated as ankyrin repeat domain-containing proteins that are similar to other NCLDV.
Among NCLDV included in this analysis, FV3 has the smallest genome and, consequently, fewer genes [25]. Of the 91 genes analyzed, 33 (36.3%) are classified as early, 22 (24.2%) as intermediate and 36 (39.5%) as late (Figure 1 D). Half of the FV3 genes have no known function. Ten genes are involved in DNA replication, recombination, and repair, with five being classified as intermediate, two as early, and three as late (Figure 2). Only four genes are involved with transcription and RNA processing, including two RNA polymerase subunits and transcription elongation factor SII, all classified as intermediate genes, and the VLTF3, expressed in the late phase of the replicative cycle. We did not observe genes involved in translation or the host-virus interaction (Figure 2). It is important to note that the absence of genes involved in direct interaction with the host as well as the fact that only a few are involved in signal transduction regulation (two early genes, four late genes) do not indicate that FV-3 only weakly manipulates the host cell. On the contrary, these data should be interpreted with caution given the large number of genes that have not been characterized, and genes involved in the manipulation of host metabolism could be identified with further studies. Curiously, most of the genes involved in virion structure, annotated as surface proteins, are classified as early genes (9/18, 50%), while only 4 (22.2%) are classified as late (Figure 2). This is in sharp contrast to other NCLDV, where genes involved in the virion structure and morphogenesis are mostly classified as intermediate or late genes. Nevertheless, the gene encoding the conserved major capsid protein is expressed late, as observed for most other NCLDV.
EhV-201 encodes 447 genes and single-cell RNA-seq data indicate that all genes are expressed throughout the replication cycle evaluated from 0 to 24 hpi [29]. These genes are expressed at different moments, which we defined as early, intermediate and late. However, due to the limited data, 52 genes (11.6%) could not be assigned to a category in the original study. Therefore, for our analysis, we considered only the genes that had been confidently classified into distinct temporal classes. From these 395 genes, 90 (22.8%) are expressed early, 185 (46.8%) are intermediate and 120 (30.4%) are late (Figure 1 E). The large majority of EhV-201 genes have no known function (374/447, 83.7%). Among the 73 genes with defined functions, 3 could not be included in any temporal class. Genes related to DNA replication, recombination, and repair are mainly expressed at intermediate moments of the replication cycle, similarly to other NCLDV, while those related to the virion structure and morphogenesis are expressed late (Figure 2). Most genes of the transcriptional apparatus of EhV-201, including all six RNA polymerase subunits, are considered intermediate genes, with only one transcription factor, VLTF2, being expressed early (Figure 2). Curiously, genes whose products are involved in lipid and protein metabolism—for example, lipases and proteases—are most expressed early, suggesting a putative role affecting the host’s metabolism.
Researchers recently evaluated the transcriptional landscape of medusavirus by using RNA-seq; this virus was isolated from hot spring water in Japan and contains 461 protein-coding genes [35]. Of these genes, 131 (28.4%) are considered early, being expressed between 0 and 2 hpi; 272 (59%) are intermediate, expressed between 2 and 4 hpi; and 58 (12.6%) are late, expressed after 4 hpi, with higher expression after 8 hpi [65] (Figure 1 F). Similar to other NCLDV, the majority of medusavirus genes are uncharacterized (359/361, 77.9%). Most genes related to DNA metabolism are expressed until 4 hpi, including DNA polymerase and viral homologs for histone proteins (Figure 2). Despite the lack of RNA polymerase, medusavirus contains some genes involved in the transcription process, such as transcription factors that are classified as intermediate genes. Interestingly, the viral poly-A polymerase is early expressed and might be related to the poly-adenylation of viral transcripts during the replication cycle. Genes involved in signal transduction are evenly distributed in the three temporal classes, suggesting a constant interaction with the metabolic pathways of the host (Figure 2). Finally, it is curious that, differently from other NCLDV, most of the genes associated with the medusavirion structure and morphogenesis are classified as intermediate genes, with only a putative membrane protein being expressed late (Figure 2). This profile differs from those observed in other NCLDV and a deeper investigation regarding the morphogenesis of this virus could bring important novelties for the field.
5. Conclusions and perspectives
Many concepts about the virosphere have changed with studies carried out over the years following the discovery of NCLDV. This group shares many genes related to the replication of the genome and the formation of the viral structure, called “viral hallmark genes”, which point to the monophyly of this group. In addition, many members of this group have a nearly complete transcriptional apparatus, which provides some independence from their hosts’ machinery. Thus, the presence of a robust transcriptional apparatus has raised much discussion about the evolutionary aspects of these viruses and their genome.
In this work, we performed a comparative analysis of groups of genes expressed at different times of infection of different members of the NCLDV group. We observed that a common characteristic of this group is a temporal expression profile of their genes throughout the replication cycle, a characteristic that has been maintained throughout the evolution. Overall, genes related to genome transcription and replication are generally expressed in the initial/middle phase of the replicative cycle, while those associated with virion morphogenesis and structure are mainly expressed in the final phase of the virus life cycle. Understanding how the genes of a given pathogen are expressed provides data that assist researchers in understanding their biology and interaction with their hosts. In addition, information regarding the regulation of the expression of these genes can also assist in studies to interrupt this process at a certain point in the cycle to contribute to the resolution of possible diseases caused by different viral pathogens. Finally, this study compiles information about the regulation of gene expression of different pathogens that opens up the field for transcription studies of other NCLDV, for which this process has not been completely elucidated. The analysis presented here provides insights into the gene expression profiles of other viral pathogens belonging to Nucleocytoviricota and can be used as a starting point for future transcriptomic investigations.
This entry is adapted from the peer-reviewed paper 10.3390/pathogens10080935
References
- Natalya Yutin; Yuri I Wolf; Didier Raoult; Eugene V Koonin; Eukaryotic large nucleo-cytoplasmic DNA viruses: Clusters of orthologous genes and reconstruction of viral genome evolution. Virology Journal 2008, 6, 223-223, 10.1186/1743-422x-6-223.
- Lakshminarayan M. Iyer; L Aravind; Eugene V. Koonin; Common Origin of Four Diverse Families of Large Eukaryotic DNA Viruses. Journal of Virology 2001, 75, 11720-11734, 10.1128/jvi.75.23.11720-11734.2001.
- Eugene V. Koonin; Natalya Yutin; Evolution of the Large Nucleocytoplasmic DNA Viruses of Eukaryotes and Convergent Origins of Viral Gigantism. Advances in Virus Research 2018, 103, 167-202, 10.1016/bs.aivir.2018.09.002.
- Julien Guglielmini; Anthony C. Woo; Mart Krupovic; Patrick Forterre; Morgan Gaia; Diversification of giant and large eukaryotic dsDNA viruses predated the origin of modern eukaryotes. Proceedings of the National Academy of Sciences 2019, 116, 19585-19592, 10.1073/pnas.1912006116.
- Eugene V. Koonin; Valerian V. Dolja; Mart Krupovic; Arvind Varsani; Yuri I. Wolf; Natalya Yutin; F. Murilo Zerbini; Jens H. Kuhn; Global Organization and Proposed Megataxonomy of the Virus World. Microbiology and Molecular Biology Reviews 2020, 84, e00061-19, 10.1128/mmbr.00061-19.
- Nadège Philippe; Matthieu Legendre; Gabriel Doutre; Yohann Couté; Olivier Poirot; Magali Lescot; Defne Arslan; Virginie Seltzer; Lionel Bertaux; Christophe Bruley; et al. Pandoraviruses: Amoeba Viruses with Genomes Up to 2.5 Mb Reaching That of Parasitic Eukaryotes. Science 2013, 341, 281-286, 10.1126/science.1239181.
- Dorine Gaëlle Reteno; Samia Benamar; Jacques Bou Khalil; Julien Andreani; Nicholas Armstrong; Thomas Klose; Michael Rossmann; Philippe Colson; Didier Raoult; Bernard La Scola; et al. Faustovirus, an Asfarvirus-Related New Lineage of Giant Viruses Infecting Amoebae. Journal of Virology 2015, 89, 6585-6594, 10.1128/jvi.00115-15.
- Leena H. Bajrai; Samia Benamar; Esam I. Azhar; Catherine Robert; Anthony Levasseur; Didier Raoult; Bernard La Scola; Kaumoebavirus, a New Virus That Clusters with Faustoviruses and Asfarviridae. Viruses 2016, 8, 278, 10.3390/v8110278.
- Julien Andreani; Sarah Aherfi; Jacques Yaacoub Bou Khalil; Fabrizio Di Pinto; Idir Bitam; Didier Raoult; Philippe Colson; Bernard La Scola; Cedratvirus, a Double-Cork Structured Giant Virus, is a Distant Relative of Pithoviruses. Viruses 2016, 8, 300, 10.3390/v8110300.
- Matthieu Legendre; Audrey Lartigue; Lionel Bertaux; Sandra Jeudy; Julia Bartoli; Magali Lescot; Jean-Marie Alempic; Claire Ramus; Christophe Bruley; Karine Labadie; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proceedings of the National Academy of Sciences 2015, 112, E5327-E5335, 10.1073/pnas.1510795112.
- M. Legendre; J. Bartoli; L. Shmakova; S. Jeudy; K. Labadie; A. Adrait; M. Lescot; O. Poirot; L. Bertaux; C. Bruley; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proceedings of the National Academy of Sciences 2014, 111, 4274-4279, 10.1073/pnas.1320670111.
- Julien Andreani; Jacques Yaacoub Bou Khalil; Madhumati Sevvana; Samia Benamar; Fabrizio Di Pinto; Idir Bitam; Philippe Colson; Thomas Klose; Michael G. Rossmann; Didier Raoult; et al. Pacmanvirus, a New Giant Icosahedral Virus at the Crossroads between Asfarviridae and Faustoviruses. Journal of Virology 2017, 91, e00212-17, 10.1128/jvi.00212-17.
- Julien Andreani; Jacques Y. B. Khalil; Emeline Baptiste; Issam Hasni; Caroline Michelle; Didier Raoult; Anthony Levasseur; Bernard La Scola; Orpheovirus IHUMI-LCC2: A New Virus among the Giant Viruses. Frontiers in Microbiology 2018, 8, 2643, 10.3389/fmicb.2017.02643.
- Genki Yoshikawa; Romain Blanc-Mathieu; Chihong Song; Yoko Kayama; Tomohiro Mochizuki; Kazuyoshi Murata; Hiroyuki Ogata; Masaharu Takemura; Medusavirus, a Novel Large DNA Virus Discovered from Hot Spring Water. Journal of Virology 2019, 93, e02130-18, 10.1128/jvi.02130-18.
- Chantal Abergel; Matthieu Legendre; Jean-Michel Claverie; The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiology Reviews 2015, 39, 779-796, 10.1093/femsre/fuv037.
- Graziele Pereira Oliveira; Ana Cláudia Dos Santos Pereira Andrade; Rodrigo Araújo Lima Rodrigues; Thalita Souza Arantes; Paulo Victor Miranda Boratto; Ludmila Karen Dos Santos Silva; Fábio Pio Dornas; Giliane De Souza Trindade; Betânia Paiva Drumond; Bernard La Scola; et al. Promoter Motifs in NCLDVs: An Evolutionary Perspective. Viruses 2017, 9, 16, 10.3390/v9010016.
- Steven S Broyles; Bruce A Knutson; Poxvirus transcription. Future Virology 2010, 5, 639-650, 10.2217/fvl.10.51.
- Sunitha Kogenaru; Yan Qing; Yinping Guo; Nian Wang; RNA-seq and microarray complement each other in transcriptome profiling. BMC Genomics 2011, 13, 629-629, 10.1186/1471-2164-13-629.
- Zhong Wang; Mark Gerstein; Michael Snyder; RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics 2008, 10, 57-63, 10.1038/nrg2484.
- Ugrappa Nagalakshmi; Karl Waern; Michael Snyder; RNA‐Seq: A Method for Comprehensive Transcriptome Analysis. Current Protocols in Molecular Biology 2009, 89, 4.11.1-4.11.13, 10.1002/0471142727.mb0411s89.
- Moss, B. Poxviridae; in Fields Virology, Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2014; pp. 2129.
- Zhilong Yang; Sara E. Reynolds; Craig A. Martens; Daniel P. Bruno; Stephen F. Porcella; Bernard Moss; Expression Profiling of the Intermediate and Late Stages of Poxvirus Replication. Journal of Virology 2011, 85, 9899-9908, 10.1128/jvi.05446-11.
- Gwenny Cackett; Dorota Matelska; Michal Sýkora; Raquel Portugal; Michal Malecki; Jürg Bähler; Linda Dixon; Finn Werner; The African Swine Fever Virus Transcriptome. Journal of Virology 2020, 94, e00119-20, 10.1128/jvi.00119-20.
- R Goorha; Frog virus 3 requires RNA polymerase II for its replication.. Journal of Virology 1980, 37, 496-9, .
- S. Majji; V. Thodima; R. Sample; D. Whitley; Y. Deng; J. Mao; V.G. Chinchar; Transcriptome analysis of Frog virus 3, the type species of the genus Ranavirus, family Iridoviridae. Virology 2009, 391, 293-303, 10.1016/j.virol.2009.06.022.
- Trevor Williams; Valérie Barbosa‐Solomieu; V. Gregory Chinchar; A Decade of Advances in Iridovirus Research. Advances in Virus Research 2004, 65, 173-248, 10.1016/s0065-3527(05)65006-3.
- J. L. Van Etten; M. V. Graves; D. G. Müller; W. Boland; N. Delaroque; Phycodnaviridae– large DNA algal viruses. Archives of Virology 2002, 147, 1479-1516, 10.1007/s00705-002-0822-6.
- W. H. Wilson; James L. Van Etten; M. J. Allen; The Phycodnaviridae: The Story of How Tiny Giants Rule the World. Current Topics in Microbiology and Immunology 2008, 328, 1-42, 10.1007/978-3-540-68618-7_1.
- Chuan Ku; Uri Sheyn; Arnau Sebé-Pedrós; Shifra Ben-Dor; Daniella Schatz; Amos Tanay; Shilo Rosenwasser; Assaf Vardi; A single-cell view on alga-virus interactions reveals sequential transcriptional programs and infection states. Science Advances 2020, 6, eaba4137, 10.1126/sciadv.aba4137.
- Bernard La Scola; Stéphane Audic; Catherine Robert; Liang Jungang; Xavier de Lamballerie; Michel Drancourt; Richard Birtles; Jean-Michel Claverie; Didier Raoult; A Giant Virus in Amoebae. Science 2003, 299, 2033-2033, 10.1126/science.1081867.
- Didier Raoult; Stéphane Audic; Catherine Robert; Chantal Abergel; Patricia Renesto; Hiroyuki Ogata; Bernard La Scola; Marie Suzan; Jean-Michel Claverie; The 1.2-Megabase Genome Sequence of Mimivirus. Science 2004, 306, 1344-1350, 10.1126/science.1101485.
- Matthieu Legendre; Sébastien Santini; Alain Rico; Chantal Abergel; Jean-Michel Claverie; Breaking the 1000-gene barrier for Mimivirus using ultra-deep genome and transcriptome sequencing. Virology Journal 2011, 8, 99-99, 10.1186/1743-422x-8-99.
- Matthieu Legendre; Stéphane Audic; Olivier Poirot; Pascal Hingamp; Virginie Seltzer; Deborah Byrne; Audrey Lartigue; Magali Lescot; Alain Bernadac; Julie Poulain; et al. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Research 2010, 20, 664-674, 10.1101/gr.102582.109.
- Rodrigo Araújo Lima Rodrigues; Amina Cherif Louazani; Agnello Picorelli; Graziele Pereira Oliveira; Francisco Pereira Lobo; Philippe Colson; Bernard La Scola; Jônatas Santos Abrahão; Analysis of a Marseillevirus Transcriptome Reveals Temporal Gene Expression Profile and Host Transcriptional Shift. Frontiers in Microbiology 2020, 11, 651, 10.3389/fmicb.2020.00651.
- Ruixuan Zhang; Hisashi Endo; Masaharu Takemura; Hiroyuki Ogata; RNA-seq of the medusavirus suggests remodeling of the host nuclear environment at an early infection stage. biorxiv 2021, , , 10.1101/2021.04.10.439121.