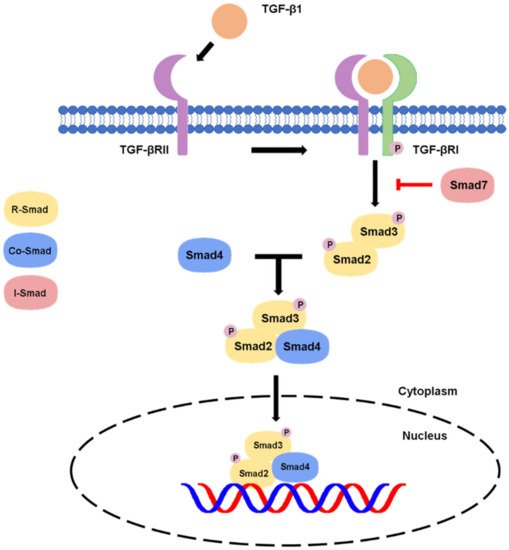
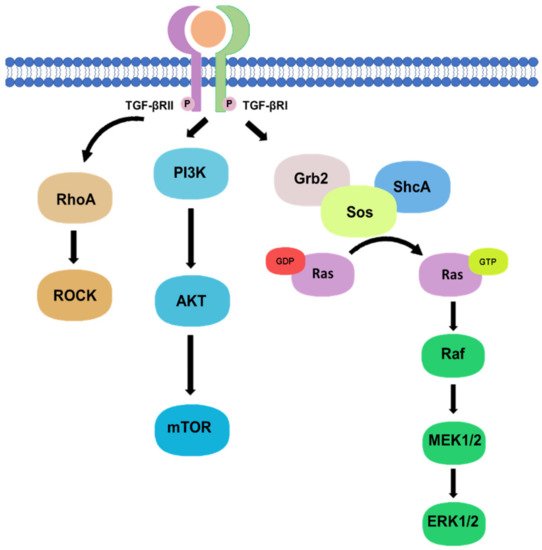
Besides, there are non-canonical pathways of the TGF-β1 signaling, where extracellular signal-regulated kinase (Erk)/mitogen-activated protein kinase (MAPK) signaling pathway are involved in the downstream signal transductions. After the phosphorylation of TGF-β type I and II receptors at tyrosine residues, adaptor proteins Src homology 2 (SH2) domain-containing protein A (ShcA) and growth factor receptor-bound protein 2 (Grb2)/son of sevenless (SOS) complex is recruited [
17]. The Grb2/SOS complex catalyzes the exchange of GDP to GTP for activating a small GTPase Ras, which then triggers the gene regulatory actions via Raf, MEK1/2, and Erk1/2 [
17,
18]. For example, Erk1/2 can phosphorylate targeted transcription factors such as Fos-related antigen 2 (Fra-2) in order to promote gene transcription [
18,
19]. In addition, protein kinase B (Akt) can be activated by TGF-β signaling via phosphatidylinositol-3 kinase (PI3K) for regulating translational responses though mTOR [
17]. Akt can also be activated in the non-canonical pathway via TRAF6-mediated Akt lysine-63 chain ubiquitination or Smad7 phosphatase and tensin homolog (PTEN) inhibition by miR-216a/217 microRNA cluster [
20,
21]. Moreover, RhoA and Rho-associated protein kinase (ROCK) can also been regulated by TGF-β signaling through a Smad independent manner [
17]. In addition, TGF-β mediates epithelial-mesenchymal transformation (EMT) of cancer cells by interfering with cell adhesion and epithelial gene expression, as well as by increasing the expression of mesenchymal proteins such as fibronectin, N-cadherin, vimentin, and fibronectin [
22]. Activation of the Smad-dependent pathway induces the expression of SNAIL, SLUG, ZEB, and TWIST transcription factors, which act as E-cadherin repressors and mediate the dissociation of desmosomes [
23]. On the other hand, activation of Smad-independent pathway promotes cytoskeletal remodelling by ERK activation and tight junction dissolution via Rho GTPase [
24]. ERK strengthens the transcriptional activity of Smad, thereby assisting TGF-β/Smad dependent EMT [
25]. The non-canonical pathways are systemically shown in
Figure 2.
Phosphorylation of TGF-β type I and II receptors can also activate downstream non-canonical pathway including Rho, PI3K/Akt, and Grb2/SOS signaling in a Smad-independent manner.