1. Overview
Many cancer studies now recognize that disease initiation, progression, and response to treatment are strongly influenced by the microenvironmental niche. Widespread desmoplasia, or fibrosis, is fundamental to pancreatic cancer development, growth, metastasis, and treatment resistance. This fibrotic landscape is largely regulated by cancer-associated fibroblasts (CAFs), which deposit and remodel extracellular matrix (ECM) in the tumor microenvironment (TME). This review will explore the prognostic and functional value of the stromal compartment in predicting outcomes and clinical prognosis in pancreatic ductal adenocarcinoma (PDAC). We will also discuss the major dynamic stromal alterations that occur in the pancreatic TME during tumor development and progression, and how the stromal ECM can influence cancer cell phenotype, metabolism, and immune response from a biochemical and biomechanical viewpoint. Lastly, we will provide an outlook on the latest clinical advances in the field of anti-fibrotic co-targeting in combination with chemotherapy or immunotherapy in PDAC, providing insight into the current challenges in treating this highly aggressive, fibrotic malignancy.
2. Pancreatic Ductal Adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer representing 95% of all patients and remains one of the most lethal forms of human cancer worldwide, with >90% of patient deaths occurring within one year of diagnosis [
1]. PDAC is projected to increase to the second-deadliest cancer type in the US by 2030, unless treatment options are improved [
1,
2]. One of the reasons PDAC has such a poor prognosis is because ~80–90% of patients first present to the clinic with advanced-stage, invasive or metastatic disease, which in most cases does not qualify for surgical removal of the tumor [
1]. As a result, surgical resection with curative intent is only available to around 10–20% of patients [
1,
3], and of those who undergo curative surgery, 80% will eventually relapse and succumb to the disease [
4]. As surgical resection is only available to a small proportion of patients, the majority of PDAC patients will be subjected to other therapies including radiation therapy, chemotherapy, chemoradiation therapy and more recently targeted therapies. For more than 20 years the standard-of-care for advanced PDAC has been gemcitabine [
5], a nucleoside analogue that inhibits DNA synthesis inducing a caspase-driven apoptotic cascade, leading to cancer cell death [
6]. Recently the addition of Abraxane (Nab-paclitaxel) to gemcitabine and the drug combination FOLFIRINOX have improved median survival in PDAC from six months (gemcitabine monotherapy) to 8.5 and 11.1 months, respectively [
7,
8,
9]. Abraxane inhibits the depolymerisation of microtubules to arrest mitosis and induce cancer cell death [
10], while FOLFIRINOX is a cocktail of cytotoxic drugs, including fluorouracil, irinotecan and oxaliplatin as well as leucovorin, which has been proven to be effective in PDAC [
7,
11]. Although FOLFIRINOX shows modest improvements in patient survival when compared with gemcitabine/Abraxane combination therapy, it also exhibits increased associated toxicities therefore mostly limiting its use to ‘fit’ patients [
12]. Thus, although significant improvements in PDAC treatment have been made, the overall five-year survival rate has remained largely unchanged for 40 years and novel therapeutics are desperately required.
PDAC is thought to arise in the ductal cells of the exocrine glands and is widely accepted as progressing through a number of pre-invasive pancreatic intraepithelial neoplasm (PanIN) stages before the development of PDAC. Several large-scale epidemiological and genetic studies have recently led to the identification of a large range of potential oncogenic drivers of the disease [
13,
14,
15]. Despite this high genetic diversity, mutation and activation of the KRAS oncogene is almost always required for the initiation of PanINs, with KRAS mutations found in 90% of all PDAC samples, driving cancer cell proliferation and survival [
16,
17,
18]. Whilst activating mutations in KRAS are responsible for initiation in the majority of PDAC cases, in vitro and in vivo studies in both human and mouse models have identified that disease progression requires subsequent mutations and/or loss of gene function such as in Trp53 (p53), SMAD4, CDKN2A and BRCA2, triggering further aberrant cell survival and proliferation, and overcoming KRAS-induced senescence [
18,
19,
20,
21,
22,
23,
24,
25,
26]. The vast heterogeneity of PDAC tumors has led to several key sequencing and proteomic studies aimed at matching individual molecular PDAC profiles with therapeutically targetable subtypes [
13,
14,
15], as previously established for other cancer types such as breast cancer.
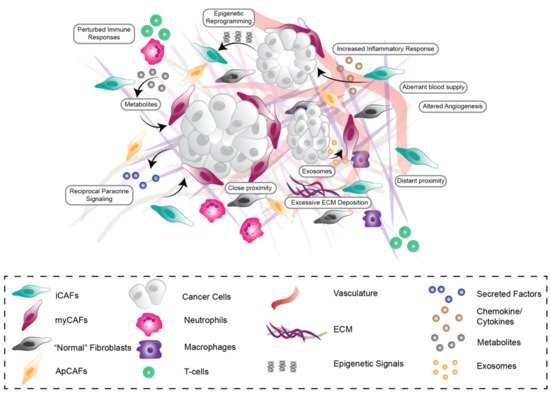
In addition to this high genetic and molecular diversity, PDAC is also one of the most stromally-dense cancer types across all malignancies, with stromal desmoplasia or fibrosis accounting for up to 90% of the total tumor volume [
27]. This desmoplasia is characterized by short and long-range reciprocal interactions between cancer cells and stromal components, including cancer-associated fibroblasts (CAFs), endothelial and immune cells as well as extracellular matrix (ECM), which influence all stages of tumorigenesis as well as therapeutic efficacy and resistance (
Figure 1) [
28,
29]. PDAC desmoplasia involves the recruitment and activation of CAFs, excessive ECM deposition, remodeling and degradation [
30,
31], increased inflammatory responses [
32], aberrant immune responses as well as altered angiogenesis and blood supply [
33,
34], which can ultimately contribute to compromised drug delivery and efficacy (
Figure 1). CAFs are one of the most abundant and active components of the PDAC microenvironment and are the main source of ECM components, such as the various types of collagens, proteoglycans, glycoproteins and hyaluronic acid (HA) [
30], which have all previously been associated with PDAC tumorigenesis. Overall, CAF-derived ECM is a dominant force in early tumor progression as well as later invasion, metastasis, and treatment resistance [
31]. The prominent role of CAFs in the microenvironment has long been assumed as an ‘Achilles’ heel’ in PDAC progression and has therefore led to numerous attempts to target them in combination with other treatment options. Surprisingly, previous work in mouse models has shown that complete depletion of stromal CAFs can lead to poorly differentiated and aggressive tumors resulting in shorter survival, indicating the stroma can also restrain cancer progression in addition to its well established pro-tumorigenic roles [
35,
36]. Additionally, it was recently reported that myofibroblast-specific deletion of Collagen I (Col I), one of the most abundant ECM proteins in PDAC, results in the acceleration of PanIN progression and PDAC emergence. Furthermore, this loss of Col I promoted an immunosuppressive TME, thereby decreasing anti-cancer immunity and overall survival [
37]. Moreover, several PDAC clinical trials reflected these unexpected pre-clinical findings where complete inhibition of stromal fibrosis via targeting of the hedgehog pathway resulted in either no added survival benefit for patients or potentially harmful side effects over gemcitabine or FOLFIRINOX alone [
38,
39,
40]. These findings suggest that a more fine-tuned, nuanced approach is required to effectively target the stroma in PDAC without causing negative side effects.
Figure 1. Stromal Heterogeneity in the pancreatic tumor microenvironment. The pancreatic tumor microenvironment is highly heterogeneous, consisting of cancer cells, activated cancer-associated fibroblast (CAF) subpopulations, increased deposition, remodeling and degradation of extracellular matrix (ECM), aberrant vasculature and impaired immune cell response. CAF subpopulations are influenced by direct-, short- and long-range growth factor (e.g., FGF1), metabolic, chemokine (e.g., CXCL12) and exosome paracrine signalling (indicated by arrows between different cell types and subpopulations) as well as epigenetic regulation via cancer cells, immune cells, vasculature and neighbouring CAFs.
CAFs are thought to originate from a diverse range of stromal cell progenitors including pancreatic stellate cells (PSCs) [
41,
42,
43,
44,
45], mesothelium [
46], resident fibroblasts [
47], mesenchymal stem cells (MSCs) [
48] and bone marrow-derived stem cells [
49]. Common “pan-CAF” markers include COL1A1/2 (collagen I alpha 1/2), DCN (decorin), PDPN (podoplanin), FAP (fibroblast activation protein) and VIM (vimentin) [
32,
50]. Recently however, the traditional view of a uniform CAF cell type within a tumor has been re-evaluated. Rather, CAFs are a highly dynamic and
heterogeneous cell population that can be both tumor-promoting and tumor-restraining [
28]. Moreover, it is now well appreciated that CAFs can rapidly respond to disruptions in tissue homeostasis, signalling and mechanical changes in their environment [
28,
29]. Recently with the advent of single cell RNA sequencing (scRNASeq), distinct subtypes of CAFs have been identified, in both mouse models of PDAC and human PDAC tissue [
32,
50,
51,
52], with new subpopulations emerging continually. In the KPC mouse model (KRAS
G12D; p53
R172H; PdxCre) and human PDAC specimens, distinct fibroblast subpopulations have been identified including myofibroblasts (myCAFs), inflammatory fibroblasts (iCAFs) and antigen presenting CAFs (apCAFs) [
32,
50] (
Figure 1). In addition to pan-CAF markers, myCAFs exhibit high expression of alpha-smooth muscle actin (α-SMA) and low levels of interleukin-6 (IL-6) as well as low levels of other inflammatory interleukins such as IL-11 and LIF (leukaemia inhibitory factor), whereas iCAFs exhibit lower levels of α-SMA and high levels of IL-6, IL-11 and LIF [
32,
50]. Meanwhile, apCAFs express pan-CAF markers as well as a range of genes relating to the MHC class II family including H2-Aa and H2-Ab1, as well as distinct pro-tumorigenic and pro-inflammatory genes such as SAA3 and SLPI [
50]. The progenitors of these CAF subpopulations are yet to be fully elucidated, however Garcia et al. (2020) recently reported that some myCAFs could be derived from GLI1+ fibroblast progenitors [
47]. CAF subpopulations also exhibit some spatial distinctions, where myCAFs tend to sequester adjacent to tumor cells and iCAFs tend to be located distally from tumor cells (
Figure 1). Moreover, it has been shown ex vivo that apCAFs could be converted to myCAFs by altering the CAF culture conditions, demonstrating that CAFs can dynamically switch subtypes according to environmental cues [
50]. This, together with the spatial organization of myCAFs and iCAFs, although not indisputable, suggests that CAFs may exhibit different phenotypes dependent on their environment, including their proximity to cancer cell stimuli. Furthermore, recent work by Feldmann et al., (2021) has shown that the transcription factor PRXX1, is in part responsible for tuning CAF activation and plasticity in PDAC tumors [
53]. In this study, deletion of PRXX1 drove the expansion of tumor-restraining CAFs, leading to increased tumor differentiation, as well as improved sensitivity to gemcitabine chemotherapy and reduced tumor dissemination [
53]. Overall, understanding more about the transcriptional and microenvironmental drivers of CAF phenotype and function in PDAC will be critical to understanding their context-dependent influence on all stages of pancreatic tumorigenesis and will inform how best to target these pro-tumorigenic features.
3. Conclusions
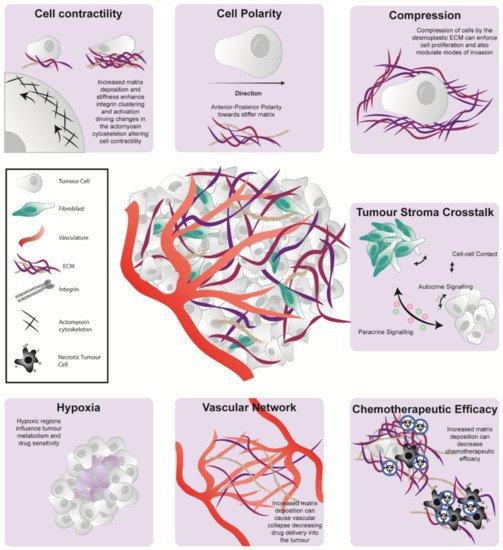
The cellular and architectural compartments of the PDAC TME play a significant role in disease development, progression, and therapeutic response (Figure 2). Here, we discussed recent studies which highlighted the unique and individual nature of the PDAC stroma and its role in contributing to tumor heterogeneity and patient prognosis. Despite the identification of several promising targets to modulate the TME, they are yet to show meaningful improvement in the clinical outcome of disease beyond early phase clinical trials. The conflicting results of several stromal targeting studies show the double-edge sword (favourable and unfavourable aspects) of stromal co-targeting; where elimination of the stromal barriers that influence the delivery of chemotherapeutic agents can also potentially drive tumor progression. As such, there is an imperative need to understand the complex role of the PDAC TME to improve stromal co-targeting regimes and enhance patient survival.
Figure 2. The pancreatic microenvironment influences tumorigenesis via a range of biochemical and biomechanical phenomena. The pancreatic TME has multiple influences on cell be havior including cell contractility (top left), cell polarity (top middle), the generation of compression forces (top right) and tumor-stroma crosstalk via direct cell-cell contact, paracrine or autocrine signaling (middle right). It can also cause the generation of hypoxic tissue pockets (bottom left), modulation of the tumor vascular network (bottom middle) and diminished cancer cell vulnerability to chemotherapy (bottom right).
This entry is adapted from the peer-reviewed paper 10.3390/cancers13143481