Primary Ciliary Dyskinesia (PCD) is a rare, under-recognized disease that affects respiratory ciliary function, resulting in chronic oto-sino-pulmonary disease. The PCD clinical phenotype overlaps with other common respiratory conditions and no single diagnostic test detects all forms of PCD. In 2018, PCD experts collaborated with the American Thoracic Society (ATS) to create a clinical diagnostic guideline for patients across North America, specifically considering the local resources and limitations for PCD diagnosis in the United States and Canada. Nasal nitric oxide (nNO) testing is recommended for first-line testing in patients ≥5 years old with a compatible clinical phenotype; however, all low nNO values require confirmation with genetic testing or ciliary electron micrograph (EM) analysis. Furthermore, these guidelines recognize that not all North American patients have access to nNO testing and isolated genetic testing is appropriate in cases with strong clinical PCD phenotypes. For unresolved diagnostic cases, referral to a PCD Foundation accredited center is recommended.
1. Recognition of the Clinical Phenotype
PCD is a disease of impaired motor cilia function associated with a growing list of causative genes. Most pediatric PCD patients exhibit several key clinical features including: (a) persistent, year-round wet cough that starts in the first year of life, (b) persistent, year-round nasal congestion that also starts in the first year of life, (c) the presence of organ laterality abnormalities, and (d) unexplained neonatal respiratory distress in infants born at term gestation [
8]. While PCD patients often display otitis media with persistent effusion, chronic bronchitis, and recurrent pneumonia, these issues are prevalent in many other children presenting for respiratory care and thus do not readily assist physicians in discerning which patients should undergo further PCD investigations. However, the complete absence of middle ear disease or recurrent lower respiratory tract infections makes PCD less likely. The ATS PCD diagnostic guidelines recommend that at least two of the key clinical PCD features be present for the pre-test probability to be high enough to proceed with PCD diagnostic testing [
3,
8].
Unlike other overlapping pediatric respiratory conditions, PCD usually presents with persistent symptoms on a daily, non-seasonal basis; the symptoms never completely resolve, even with prolonged antibiotic courses. The persistent wet cough and rhinorrhea often appear before 6 months of age, though many families report onset at birth. Neonatal respiratory distress, despite term birth, is seen in at least 80% of PCD cases and can have a delayed onset (median onset at 12 h of life, with a range of 0 to 72 h of life), accompanied by lobar atelectasis on chest radiography [
9]. The duration of neonatal respiratory distress in PCD is often prolonged, with a median of 14 days [
8,
9], and some cases require supplemental oxygen therapy for several months.
In adult PCD populations, persistent wet cough and nasal congestion on a daily basis remain key clinical features, though the age of symptom onset and presence of neonatal respiratory distress are often forgotten in these cases [
10]. Chronic sinusitis with polyposis and male infertility, as well as female subfertility, are also frequently reported by adults with PCD; additionally, bronchiectasis is universally present in the adult PCD populations.
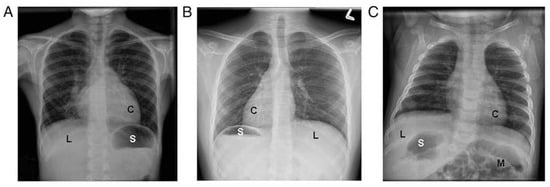
Organ laterality defects are also common in PCD, due to the motility defects of the embryonic monocilia, which appear during early embryogenesis. However, organ laterality defects only occur in 50% of individuals with PCD and can result in a variety of organ arrangements, including situs inversus totalis (mirror image organ arrangement), situs ambiguus (a mix or left-right organ arrangement, often with congenital heart defects), isolated organ laterality defects (such as dextrocardia), polysplenia, interrupted inferior vena cava, and others [
11] (
Figure 1). When situs ambiguus occurs with complex congenital heart lesions, the label “heterotaxy” is often used, though situs ambiguus organ arrangements can similarly occur with mild congenital heart defects (such as ventricular septal defects). The terms situs ambiguus and heterotaxy are often employed interchangeably, which may result in confusion amongst clinicians.
Figure 1. Examples of laterality defects on radiology imaging: (
A) situs solitus, (
B) situs inversus totalis, and (
C) situs ambiguus. (C = cardiac apex; L = liver; M = intestinal malrotation; S = stomach). Reprinted with permission from ref. [
11]. Copyright 2014 Shapiro AJ, Davis SD, Ferkol T, Dell SD, Rosenfeld M, Olivier KN, et al.
2. PCD Diagnostic Testing
2.1. Nasal NO Testing
Nitric oxide (NO) is a colorless, odorless gas produced in the upper and lower respiratory epithelium. It has diverse effects in the respiratory system, including vasodilation, bacterial killing, and the modulation of inflammation [
12,
13]. Fractional exhaled NO from the lower airways (FeNO) is normally upregulated in times of infection and inflammation (as often seen in poorly controlled asthma), while nasal NO is mainly produced in the paranasal sinuses.
Nasal nitric oxide (nNO) measurement represents a simple, non-invasive way to screen patients for PCD, with results immediately available at the time of testing. For unclear reasons, nNO values are distinctly reduced in individuals with PCD [
14]. Other conditions that may have overlapping low nNO values include cystic fibrosis, diffuse pan-bronchiolitis, acute viral respiratory infection, and certain forms of primary immunodeficiency [
15,
16,
17,
18,
19,
20]. Thus, these conditions should always be considered and investigated if relying on low nNO as the sole diagnostic evidence for likely PCD (i.e., negative genetic testing and normal TEM analysis). When nNO values are repeated over time and remain persistently low, they can yield a provisional diagnosis of PCD, but cannot fully confirm the diagnosis and must be taken in context with other PCD diagnostic test results. It should be noted that only chemiluminescence NO devices have been prospectively validated for use in PCD investigations, with results from significantly less expensive electrochemical NO devices lacking robust studies in large PCD populations [
21,
22].
Cross-sectional and longitudinal observations have shown that ≥90% of individuals with PCD have nasal NO levels below 77 nL/min [
18], when measured per a standardized protocol. Pooled sensitivity and specificity estimates for nNO < 77 nL/min are 96% and 96%, respectively, in cases of PCD that were confirmed using TEM and/or genetic analysis and when cystic fibrosis has been ruled out [
23]. Low nasal NO values should be confirmed with at least one additional test on a separate visit, with patients at their baseline health, and without evidence of viral respiratory infection.
Around 5–10% of PCD cases have nNO values above the established cut-off of 77 nL/min, specifically those associated with mutations in radial spoke or central apparatus proteins [
23,
24]. These include cases due to genetic variants in
RSPH1, which are associated with normal ciliary ultrastructure by TEM and nNO values of 100 to 300 nL/min [
18] (
Table 1).
Table 1. PCD genotypes grouped by reported nasal nitric oxide (nNO) values. Reported case numbers of specific genes with nNO > 77nl/min are presented in parentheses.
| nNO Routinely ≤ 77 nL/min |
Limited Cases with nNO > 77 nL/min |
nNO Routinely > 77 nL/min |
| ARMC4 |
DNAI2 |
DNAH9 (9 cases) [25,26] |
CCDC103 [27] |
| CCDC39 |
DNAJB13 |
TTC12 (2 cases) [28] |
RSPH1 [29] |
| CCDC40 |
DNAL1 |
RPGR (<10 cases) [30,31] |
|
| CCDC114 |
DRC1 |
CCNO (2 cases) [32] |
|
| CCDC151 |
DRC2 |
FOXJ1 (4 cases) [33] |
|
| CFAP57 |
HYDIN |
NEK10 (1 case) [34,35] |
|
| CFAP298 |
LRRC6 |
GAS2L2 (2 cases) [36] |
|
| CFAP300 |
MCIDAS |
GAS8 (1 cases) [37] |
|
| DNAAF1 |
NME5 |
STK36 (1 case) [38] |
|
| DNAAF2 |
NME8 |
CFAP221 (3 cases) [39] |
|
| DNAAF3 |
OFD1 |
SPEF2 (1 cases) [40,41,42] |
|
| DNAAF4 |
PIHID3 |
LRRC56 (1 case) [43] |
|
| DNAAF5 |
RSPH3 |
|
|
| DNAH1 |
RSPH4A |
|
|
| DNAH5 |
RSPH9 |
|
|
| DNAH8 |
SPAG1 |
|
|
| DNAH11 |
TTC25 |
|
|
| DNAI1 |
ZMYND10 |
Successful nNO testing requires a patient be cooperative enough to blow into a resistor device and achieve steady state NO measurement. Children under 5 years old often cannot perform this maneuver, so nNO testing may not be appropriate in preschool-aged children. Tidal breathing nNO measurements for patients 2–5 years old are feasible and are currently under investigation; however, diagnostic cut-off values are undefined in this age group. According to ATS guidelines, in patients with an appropriate PCD clinical phenotype and one nNO value <77 nL/min, a repeat measurement is recommended at least 2 weeks later [
44]. With repeat low nNO values and negative cystic fibrosis testing, a presumptive diagnosis of PCD can be made and appropriate PCD therapies commenced. However, the availability of nNO testing is still hampered by the lack of clinical regulatory approval in North America; thus, most nNO tests are performed under research protocols in certified PCD centers, which are not reimbursed by insurance plans.
2.2. PCD Genetic Testing
PCD is a genetically heterogenous disease caused by variants of more than 50 genes. This number will probably increase as more gene mutations are associated with PCD. The most frequently reported mutations occur in
DNAI1 and
DNAH5, which were the first implicated PCD genes [
45,
46]. The high number of pathogenic variants in different genes associated with PCD is a derivative of the complexity of the motile cilium, which is composed of hundreds of structural proteins and an unknown number of cytoplasmic factors that are required for ciliary assembly. Most PCD gene mutations are inherited in an autosomal recessive fashion. However, other inheritance modes occur, including x-linked inheritance in the assembly factor
PIH1D3 and autosomal dominant inheritance in mutations of the forked head transcription factor
FOXJ1 [
33,
47,
48]. It is believed that mutations in known PCD-associated genes account for approximately 70–80% of PCD cases [
49] (
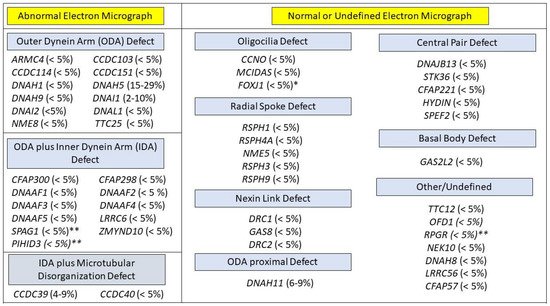
Figure 2).
Figure 2. PCD genotypes, grouped by expected ciliary ultrastructure on electron micrograph (EM). Genotypes with an x-linked mode of inheritance are labeled with **. Genotypes in with an autosomal dominant mode of inheritance are labled with *. All other genotypes have an autosomal recessive mode of inheritance. Approximate prevalence of each genotype appears in paretheses.
Numerous commercial PCD genetic panels are available, varying in the number of genes covered and the employed sequencing technology. Most panels include the next generation sequencing analysis of at least 30 genes, with some offering more than 40 genes, plus deletion/duplication analysis. Panels covering greater numbers of PCD genes will have greater detection rates [
50] (
Figure 2). It is expected that the routine use of newer sequencing methods, such as whole exome sequencing (which offers better coverage), will likely replace panels with limited numbers of genes [
51].
Although the increased availability of genetic tools is transformative for PCD diagnosis, interpreting the results of these tests may be challenging, due to the high number of variants of unknown significance (VUS), which often occur when there is discrepancy in the prediction tools used to determine pathogencity or when mutations were not previously reported to be pathogenic. PCD diagnosis requires two pathogenic variants be found in a single known PCD gene, on two opposite chromosomes (in trans) or on one allele, in the case of x-linked or autosomal dominant forms (PIH1D3, RPGR, OFD1, or FOXJ1). Pathogenicity of VUS results should be evaluated on a case-by-case basis, after consultation with either a geneticist or a PCD specialty center; VUS results cannot be assumed as disease-causing for diagnostic purposes.
2.3. Cilia Ultrastructure Evaluation
Traditionally, the use of TEM to analyze ciliary ultrastructure was considered the gold standard for a diagnosis of PCD. Motile ciliary cross sections show a typical 9 + 2 arrangement (
Figure 2) that represents 9 outer microtubule doublets surrounding a central pair. Outer dynein arms (ODA) and inner dynein arms (IDA) extend from the microtubule, acting as the motors powering ciliary movement. [
52]. Radial spokes and nexin links provide stability to beating ciliary axonemes.
Procuring and processing TEM samples requires significant expertise. From sample collection considerations (i.e., nasal versus endobronchial scraping as opposed to “pinch” tracheal biopsies) to sample processing considerations (i.e., choice of fixatives and skill of technicians in choosing multiple ciliary tufts across the sample) the TEM image quality can vary greatly. Interpretation is similarly challenging and requires evaluation by pathologists with extensive experience examining ciliary ultrastructure. To address these challenges, a more recent international consensus guideline focused on reporting TEM results in the diagnosis of PCD, which included minimal criteria for acceptable TEM studies and the creation of various diagnostic classes of TEM defects. Per this protocol, only “hallmark” defects (class 1: absent ODA, absent ODA+IDA, and absent IDA with microtubular disorganization) are reliably diagnostic of PCD [
53]. Ultimately, 30% of genetically proven cases of PCD do not have a hallmark defect on TEM [
1], due to the limited sensitivity of EM in identifying small structural changes in the electron dense ciliary axoneme. This is especially true when identifying changes affecting the central pair, radial spokes, or inner dynein arms [
54].
A promising diagnostic test using immunofluorescent-labeled antibody microscopy has been adopted in some European PCD centers [
55]. Immunofluorescent antibody microscopy has the advantage of being more readily available than TEM, costing less per sample, and is possibly more sensitive than TEM depending on the panel of antibodies used. However, the diagnostic accuracy of immunofluorescent antibody testing has not been firmly established in PCD [
55]. Most local pathology laboratories also possess the technical knowledge to process and interpret immunofluorescence antibody samples, as this technique is currently used in a variety of human diseases. Immunofluorescent antibody microscopy is currently not available for clinical use in North American centers, though this technique is being considered at several centers.
This entry is adapted from the peer-reviewed paper 10.3390/diagnostics11071278