Mitochondrial dysfunction is known to contribute to mitochondrial diseases, as well as to a variety of aging-based pathologies. Mitochondria have their own genomes (mitochondrial DNA (mtDNA)) and the abnormalities, such as point mutations, deletions, and copy number variations, are involved in mitochondrial dysfunction. In recent years, several epidemiological studies and animal experiments have supported the Developmental Origin of Health and Disease (DOHaD) theory, which states that the environment during fetal life influences the predisposition to disease and the risk of morbidity in adulthood. Mitochondria play a central role in energy production, as well as in various cellular functions, such as apoptosis, lipid metabolism, and calcium metabolism.
- DOHaD
- environmental stress
- mitochondrial DNA copy number
- pregnancy
- risk management
1. Overview
Mitochondrial dysfunction is known to contribute to mitochondrial diseases, as well as to a variety of aging-based pathologies. Mitochondria have their own genomes (mitochondrial DNA (mtDNA)) and the abnormalities, such as point mutations, deletions, and copy number variations, are involved in mitochondrial dysfunction. In recent years, several epidemiological studies and animal experiments have supported the Developmental Origin of Health and Disease (DOHaD) theory, which states that the environment during fetal life influences the predisposition to disease and the risk of morbidity in adulthood. Mitochondria play a central role in energy production, as well as in various cellular functions, such as apoptosis, lipid metabolism, and calcium metabolism. In terms of the DOHaD theory, mtDNA copy number may be a mediator of health and disease. This paper summarizes the results of recent epidemiological studies on the relationship between environmental factors and mtDNA copy number during pregnancy from the perspective of DOHaD theory. The results of these studies suggest a hypothesis that mtDNA copy number may reflect environmental influences during fetal life and possibly serve as a surrogate marker of health risks in adulthood.
2. Mitochondrial Diseases
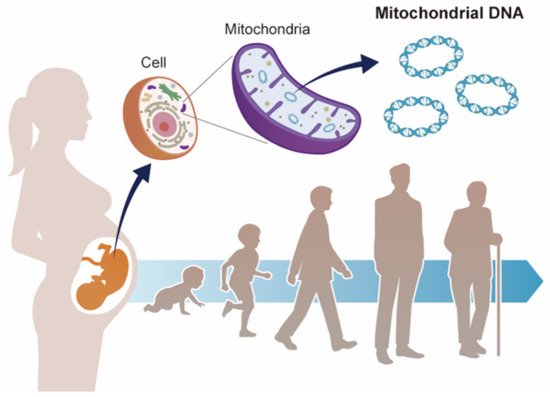
3. DOHaD Theory
This entry is adapted from the peer-reviewed paper 10.3390/ijms22126634
References
- Luft, R.; Ikkos, D.; Palmieri, G.; Ernster, L.; Afzerius, B. A case of severe hypermetabolism of nonthyroid origin with a defect in the maintenance of mitochondrial respiratory control: A correlated clinical, biochemical, and morphological study. J. Clin. Investig. 1962, 41, 1776–1804.
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192.
- Brillo, V.; Chieregato, L.; Leanza, L.; Muccioli, S.; Costa, R. Mitochondrial Dynamics, ROS, and Cell Signaling: A Blended Overview. Life 2021, 11, 332.
- Ng, Y.S.; Turnbull, D.M. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191.
- Gibson, K.; Halliday, J.L.; Kirby, D.M.; Yaplito-Lee, J.; Thorburn, D.R.; Boneh, A. Mitochondrial oxidative phosphorylation disorders presenting in neonates: Clinical manifestations and enzymatic and molecular diagnoses. Pediatrics 2008, 122, 1003–1008.
- Ohtake, A.; Murayama, K.; Mori, M.; Harashima, H.; Yamazaki, T.; Tamaru, S.; Yamashita, Y.; Kishita, Y.; Nakachi, Y.; Kohda, M.; et al. Diagnosis and molecular basis of mitochondrial respiratory chain disorders: Exome sequencing for disease gene identification. Biochim. Biophys. Acta 2014, 1840, 1355–1359.
- Kohda, M.; Tokuzawa, Y.; Kishita, Y.; Nyuzuki, H.; Moriyama, Y.; Mizuno, Y.; Hirata, T.; Yatsuka, Y.; Yamashita-Sugahara, Y.; Nakachi, Y.; et al. A Comprehensive Genomic Analysis Reveals the Genetic Landscape of Mitochondrial Respiratory Chain Complex Deficiencies. PLoS Genet. 2016, 12, e1005679.
- Pavlakis, S.G.; Phillips, P.C.; DiMauro, S.; de Vivo, D.C.; Rowland, L.P. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: A distinctive clinical syndrome. Ann. Neurol. 1984, 16, 481–488.
- Goto, Y.I.; Nonaka, I.; Horai, S. A mutation in the tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature 1990, 348, 651–653.
- Yatsuga, S.; Povalko, N.; Nishioka, J.; Katayama, K.; Kakimoto, N.; Matsuishi, T.; Kakuma, T.; Koga, Y. MELAS: A nationwide prospective cohort study of 96 patients in Japan. Biochim. Biophys. Acta Gen. Subj. 2012, 1820, 619–624.
- Fukuhara, N.; Tokiguchi, S.; Shirakawa, K.; Tsubaki, T. Myoclonus epilepsy associated with ragged-red fibres (mitochondrial abnormalities): Disease entity or a syndrome? Light- and electron-microscopic studies of two cases and review of literature. J. Neurol. Sci. 1980, 47, 117–133.
- Shoffner, J.M.; Lott, M.T.; Lezza, A.M.S.; Seibel, P.; Ballinger, S.W.; Wallace, D.C. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNALys mutation. Cell 1990, 61, 931–937.
- Holt, I.J.; Harding, A.E.; Cooper, J.M.; Schapira, A.H.V.; Toscano, A.; Clark, J.B.; Morgan-Hughes, J.A. Mitochondrial myopathies: Clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann. Neurol. 1989, 26, 699–708.
- Moraes, C.T.; DiMauro, S.; Zeviani, M.; Lombes, A.; Shanske, S.; Miranda, A.F.; Nakase, H.; Bonilla, E.; Werneck, L.C.; Servidei, S.; et al. Mitochondrial DNA Deletions in Progressive External Ophthalmoplegia and Kearns-Sayre Syndrome. N. Engl. J. Med. 1989, 320, 1293–1299.
- Khambatta, S.; Nguyen, D.L.; Beckman, T.J.; Wittich, C.M. Kearns-Sayre syndrome: A case series of 35 adults and children. Int. J. Gen. Med. 2014, 7, 325–332.
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- John, J.C.S. Genomic Balance: Two Genomes Establishing Synchrony to Modulate Cellular Fate and Function. Cells 2019, 8, 1306.
- Al Rawi, S.; Louvet-Vallée, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147.
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144.
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020, 219, e202004029.
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705.
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762.
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612.
- Pohjoismäki, J.L.O.; Goffart, S.; Tyynismaa, H.; Willcox, S.; Ide, T.; Kang, D.; Suomalainen, A.; Karhunen, P.J.; Griffith, J.D.; Holt, I.J.; et al. Human heart mitochondrial DNA is organized in complex catenated networks containing abundant four-way junctions and replication forks. J. Biol. Chem. 2009, 284, 21446–21457.
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658.
- Barchiesi, A.; Vascotto, C. Transcription, processing, and decay of mitochondrial RNA in health and disease. Int. J. Mol. Sci. 2019, 20, 2221.
- Wei, W.; Tuna, S.; Keogh, M.J.; Smith, K.R.; Aitman, T.J.; Beales, P.L.; Bennett, D.L.; Gale, D.P.; Bitner-Glindzicz, M.A.K.; Black, G.C.; et al. Germline selection shapes human mitochondrial DNA diversity. Science 2019, 364, eaau6520.
- Schaefer, A.M.; Taylor, R.W.; Turnbull, D.M.; Chinnery, P.F. The epidemiology of mitochondrial disorders—Past, present and future. Biochim. Biophys. Acta 2004, 1659, 115–120.
- Schaefer, A.M.; McFarland, R.; Blakely, E.L.; He, L.; Whittaker, R.G.; Taylor, R.W.; Chinnery, P.F.; Turnbull, D.M. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 2008, 63, 35–39.
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759.
- Murayama, K.; Shimura, M.; Liu, Z.; Okazaki, Y.; Ohtake, A. Recent topics: The diagnosis, molecular genesis, and treatment of mitochondrial diseases. J. Hum. Genet. 2019, 64, 113–125.
- Frazier, A.E.; Thorburn, D.R.; Compton, A.G. Mitochondrial energy generation disorders: Genes, mechanisms, and clues to pathology. J. Biol. Chem. 2019, 294, 5386–5395.
- Sato, A.; Kono, T.; Nakada, K.; Ishikawa, K.; Inoue, S.I.; Yonekawa, H.; Hayashi, J.I. Gene therapy for progeny of mito-mice carrying pathogenic mtDNA by nuclear transplantation. Proc. Natl. Acad. Sci. USA 2005, 102, 16765–16770.
- Yamada, Y.; Harashima, H. Mitochondrial drug delivery systems for macromolecule and their therapeutic application to mitochondrial diseases. Adv. Drug Deliv. Rev. 2008, 60, 1439–1462.
- Rai, P.K.; Craven, L.; Hoogewijs, K.; Russell, O.M.; Lightowlers, R.N. Advances in methods for reducing mitochondrial DNA disease by replacing or manipulating the mitochondrial genome. Essays Biochem. 2018, 62, 455–465.
- Yamada, Y.; Ito, M.; Arai, M.; Hibino, M.; Tsujioka, T.; Harashima, H. Challenges in promoting mitochondrial transplantation therapy. Int. J. Mol. Sci. 2020, 21, 6365.
- Park, A.; Oh, M.; Lee, S.J.; Oh, K.-J.; Lee, E.-W.; Lee, S.C.; Bae, K.-H.; Han, B.S.; Kim, W.K. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793.
- Mengel-From, J.; Thinggaard, M.; Dalgård, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014, 133, 1149–1159.
- Ashar, F.N.; Moes, A.; Moore, A.Z.; Grove, M.L.; Chaves, P.H.M.; Coresh, J.; Newman, A.B.; Matteini, A.M.; Bandeen-Roche, K.; Boerwinkle, E.; et al. Association of mitochondrial DNA levels with frailty and all-cause mortality. J. Mol. Med. 2015, 93, 177–186.
- Dolcini, J.; Kioumourtzoglou, M.-A.; Cayir, A.; Sanchez-Guerra, M.; Brennan, K.J.; Dereix, A.E.; Coull, B.A.; Spiro, A.; Vokonas, P.; Schwartz, J.; et al. Age and mitochondrial DNA copy number influence the association between outdoor temperature and cognitive function. Environ. Epidemiol. 2020, 4, e0108.
- Castellani, C.A.; Longchamps, R.J.; Sumpter, J.A.; Newcomb, C.E.; Lane, J.A.; Grove, M.L.; Bressler, J.; Brody, J.A.; Floyd, J.S.; Bartz, T.M.; et al. Mitochondrial DNA copy number can influence mortality and cardiovascular disease via methylation of nuclear DNA CpGs. Genome Med. 2020, 12, 84.
- Fazzini, F.; Lamina, C.; Fendt, L.; Schultheiss, U.T.; Kotsis, F.; Hicks, A.A.; Meiselbach, H.; Weissensteiner, H.; Forer, L.; Krane, V.; et al. Mitochondrial DNA copy number is associated with mortality and infections in a large cohort of patients with chronic kidney disease. Kidney Int. 2019, 96, 480–488.
- Spinazzola, A.; Invernizzi, F.; Carrara, F.; Lamantea, E.; Donati, A.; DiRocco, M.; Giordano, I.; Meznaric-Petrusa, M.; Baruffini, E.; Ferrero, I.; et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J. Inherit. Metab. Dis. 2009, 32, 143–158.
- Copeland, W.C. Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 64–74.
- Young, M.J.; Copeland, W.C. Human mitochondrial DNA replication machinery and disease. Curr. Opin. Genet. Dev. 2016, 38, 52–62.
- Suomalainen, A.; Isohanni, P. Mitochondrial DNA depletion syndromes—Many genes, common mechanisms. Neuromuscul. Disord. 2010, 20, 429–437.
- Sun, X.; John, J.C.S. The role of the mtDNA set point in differentiation, development and tumorigenesis. Biochem. J. 2016, 473, 2955–2971.
- Gemma, C.; Sookoian, S.; Alvariñas, J.; García, S.I.; Quintana, L.; Kanevsky, D.; González, C.D.; Pirola, C.J. Mitochondrial DNA depletion in small- and large-for-gestational-age newborns. Obesity 2006, 14, 2193–2199.
- Barker, D.J.P.; Godfrey, K.M.; Fall, C.; Osmond, C.; Winter, P.D.; Shaheen, S.O. Relation of birth weight and childhood respiratory infection to adult lung function and death from chronic obstructive airways disease. BMJ 1991, 303, 671–675.
- Ramadhani, M.K.; Grobbee, D.E.; Bots, M.L.; Cabezas, M.C.; Vos, L.E.; Oren, A.; Uiterwaal, C.S.P.M. Lower birth weight predicts metabolic syndrome in young adults: The Atherosclerosis Risk in Young Adults (ARYA)-study. Atherosclerosis 2006, 184, 21–27.
- Geelhoed, J.J.M.; Jaddoe, V.W.V. Early influences on cardiovascular and renal development. Eur. J. Epidemiol. 2010, 25, 677–692.
- Memon, A.A.; Sundquist, J.; Hedelius, A.; Palmér, K.; Wang, X.; Sundquist, K. Association of mitochondrial DNA copy number with prevalent and incident type 2 diabetes in women: A population-based follow-up study. Sci. Rep. 2021, 11, 4608.
- Yang, S.Y.; Castellani, C.A.; Longchamps, R.J.; Pillalamarri, V.K.; O’Rourke, B.; Guallar, E.; Arking, D.E. Blood-derived mitochondrial DNA copy number is associated with gene expression across multiple tissues and is predictive for incident neurodegenerative disease. Genome Res. 2021, 31, 349–358.
- Barker, D.J.P. The fetal and infant origins of adult disease. BMJ 1990, 301, 1111.
- Gluckman, P.D.; Hanson, M.A. Living with the past: Evolution, development, and patterns of disease. Science 2004, 305, 1733–1736.
- Baird, J.; Jacob, C.; Barker, M.; Fall, C.; Hanson, M.; Harvey, N.; Inskip, H.; Kumaran, K.; Cooper, C. Developmental Origins of Health and Disease: A Lifecourse Approach to the Prevention of Non-Communicable Diseases. Healthcare 2017, 5, 14.
- Frankenhuis, W.E.; Nettle, D.; McNamara, J.M. Echoes of Early Life: Recent Insights From Mathematical Modeling. Child Dev. 2018, 89, 1504–1518.
- Goyal, D.; Limesand, S.W.; Goyal, R. Epigenetic responses and the developmental origins of health and disease. J. Endocrinol. 2019, 242, T105–T119.
- Penkler, M.; Hanson, M.; Biesma, R.; Müller, R. DOHaD in science and society: Emergent opportunities and novel responsibilities. J. Dev. Orig. Health Dis. 2019, 10, 268–273.
- Fall, C.H.D.; Kumaran, K. Metabolic programming in early life in humans. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180123.
- Briana, D.D.; Malamitsi-Puchner, A. Perinatal biomarkers implying “Developmental Origins of Health and Disease” consequences in intrauterine growth restriction. Acta Paediatr. Int. J. Paediatr. 2020, 109, 1317–1322.
- Vermeulen, R.; Schymanski, E.L.; Barabási, A.L.; Miller, G.W. The exposome and health: Where chemistry meets biology. Science 2020, 367, 392–396.
- Barker, D.J.P.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 327, 1077–1081.
- Barker, D.J.P.; Osmond, C.; Winter, P.D.; Margetts, B.; Simmonds, S.J. Weight in infancy and death from ischaemic heart disease. Lancet 1989, 334, 577–580.
- Barker, D.J.P.; Bull, A.R.; Osmond, C.; Simmonds, S.J. Fetal and placental size and risk of hypertension in adult life. BMJ 1990, 301, 259–262.
- Barker, D.J.P.; Hales, C.N.; Fall, C.H.D.; Osmond, C.; Phipps, K.; Clark, P.M.S. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): Relation to reduced fetal growth. Diabetologia 1993, 36, 62–67.
- Hofman, P.L.; Regan, F.; Jackson, W.E.; Jefferies, C.; Knight, D.B.; Robinson, E.M.; Cutfield, W.S. Premature Birth and Later Insulin Resistance. N. Engl. J. Med. 2004, 351, 2179–2186.
- Barker, D.J.P. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417.
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491.
- Eriksson, J.G. Developmental Origins of Health and Disease—From a small body size at birth to epigenetics. Ann. Med. 2016, 48, 456–467.
- Hanson, M. The birth and future health of DOHaD. J. Dev. Orig. Health Dis. 2015, 6, 434–437.
- Hsu, C.N.; Lin, Y.J.; Yu, H.R.; Lin, I.C.; Sheen, J.M.; Huang, L.T.; Tain, Y.L. Protection of male rat offspring against hypertension programmed by prenatal dexamethasone administration and postnatal high-fat diet with the Nrf2 activator dimethyl fumarate during pregnancy. Int. J. Mol. Sci. 2019, 20, 3957.
- Hsu, C.N.; Lin, Y.J.; Tain, Y.L. Maternal exposure to bisphenol a combined with high-fat diet-induced programmed hypertension in adult male rat offspring: Effects of resveratrol. Int. J. Mol. Sci. 2019, 20, 4382.
- Larsen, T.D.; Sabey, K.H.; Knutson, A.J.; Gandy, T.C.T.; Louwagie, E.J.; Lauterboeck, L.; Mdaki, K.S.; Baack, M.L. Diabetic pregnancy and maternal high-fat diet impair mitochondrial dynamism in the developing fetal rat heart by sex-specific mechanisms. Int. J. Mol. Sci. 2019, 20, 3090.
- Hofstee, P.; Cuffe, J.S.M.; Perkins, A.V. Analysis of selenoprotein expression in response to dietary selenium deficiency during pregnancy indicates tissue specific differential expression in mothers and sex specific changes in the fetus and offspring. Int. J. Mol. Sci. 2020, 21, 2210.
- Alexandre-Gouabau, M.C.; David-Sochard, A.; Royer, A.L.; Parnet, P.; Paillé, V. Moderate high caloric maternal diet impacts dam breast milk metabotype and offspring lipidome in a sex-specific manner. Int. J. Mol. Sci. 2020, 21, 5428.
- Visker, J.R.; Dangott, L.J.; Leszczynski, E.C.; Ferguson, D.P. Postnatal growth restriction in mice alters cardiac protein composition and leads to functional impairment in adulthood. Int. J. Mol. Sci. 2020, 21, 9459.
- Hales, C.N.; Barker, D.J.P. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20.
- Heindel, J.J.; Skalla, L.A.; Joubert, B.R.; Dilworth, C.H.; Gray, K.A. Review of developmental origins of health and disease publications in environmental epidemiology. Reprod. Toxicol. 2017, 68, 34–48.
- Almeida, D.L.; Pavanello, A.; Saavedra, L.P.; Pereira, T.S.; de Castro-Prado, M.A.A.; de Freitas Mathias, P.C. Environmental monitoring and the developmental origins of health and disease. J. Dev. Orig. Health Dis. 2019, 10, 608–615.
- Street, M.E.; Bernasconi, S. Endocrine-disrupting chemicals in human fetal growth. Int. J. Mol. Sci. 2020, 21, 1430.
- Chen, R.; Clifford, A.; Lang, L.; Anstey, K.J. Is exposure to secondhand smoke associated with cognitive parameters of children and adolescents?—A systematic literature review. Ann. Epidemiol. 2013, 23, 652–661.
- Brenner, B.M.; Chertow, G.M. Congenital Oligonephropathy and the Etiology of Adult Hypertension and Progressive Renal Injury. Am. J. Kidney Dis. 1994, 23, 171–175.
- Tain, Y.L.; Huang, L.T.; Hsu, C.N. Developmental programming of adult disease: Reprogramming by melatonin? Int. J. Mol. Sci. 2017, 18, 426.
- Hsu, C.N.; Tain, Y.L. Light and circadian signaling pathway in pregnancy: Programming of adult health and disease. Int. J. Mol. Sci. 2020, 21, 2232.
- Iwama, N.; Metoki, H.; Ohkubo, T.; Ishikuro, M.; Obara, T.; Kikuya, M.; Yagihashi, K.; Nishigori, H.; Sugiyama, T.; Sugawara, J.; et al. Maternal clinic and home blood pressure measurements during pregnancy and infant birth weight: The BOSHI study. Hypertens. Res. 2016, 39, 151–157.
- Aisa, M.C.; Cappuccini, B.; Barbati, A.; Clerici, G.; Torlone, E.; Gerli, S.; Di Renzo, G.C. Renal Consequences of Gestational Diabetes Mellitus in Term Neonates: A Multidisciplinary Approach to the DOHaD Perspective in the Prevention and Early Recognition of Neonates of GDM Mothers at Risk of Hypertension and Chronic Renal Diseases in Later Life. J. Clin. Med. 2019, 8, 429.
- Gluckman, P.D.; Hanson, M.A.; Buklijas, T.; Low, F.M.; Beedle, A.S. Epigenetic mechanisms that underpin metabolic and cardiovascular diseases. Nat. Rev. Endocrinol. 2009, 5, 401–408.
- Chmurzynska, A. Fetal programming: Link between early nutrition, DNA methylation, and complex diseases. Nutr. Rev. 2010, 68, 87–98.
- Safi-Stibler, S.; Gabory, A. Epigenetics and the Developmental Origins of Health and Disease: Parental environment signalling to the epigenome, critical time windows and sculpting the adult phenotype. Semin. Cell Dev. Biol. 2020, 97, 172–180.
- Sarkar, A.; Yoo, J.Y.; Valeria Ozorio Dutra, S.; Morgan, K.H.; Groer, M. The Association between Early-Life Gut Microbiota and Long-Term Health and Diseases. J. Clin. Med. 2021, 10, 459.
- Rodriguez-Caro, H.; Williams, S.A. Strategies to reduce non-communicable diseases in the offspring: Negative and positive in utero programming. J. Dev. Orig. Health Dis. 2018, 9, 642–652.
- Tu’akoi, S.; Vickers, M.H.; Bay, J.L. DOHaD in low- and middle-income countries: A systematic review exploring gaps in DOHaD population studies. J. Dev. Orig. Health Dis. 2020, 11, 557–563.