Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Pediatrics
The mammalian target of the rapamycin (mTOR) system regulates various cellular functions, such as growth, proliferation, metabolism and survival/death. In systemic organs, it is critically involved in multiple processes, including neurogenesis, nutrition and immunity. In the brain, its roles are essential in the cerebral cortical development, synaptic functions, and brain activities such as learning, cognition and social functions.
- mTORopathy
- mTOR inhibitor
- TSC
- epilepsy
- intellectual disability
- autism
- epileptic encephalopathy
1. Introduction
The mammalian target of the rapamycin (mTOR) system is an essential signal transduction system inherent in all mammalian cells [1] (Figure 1). Its upstream area consists of several branches. One branch receives extracellular signals, such as insulin and insulin-like growth factors (IGFs), then transmits the information via phosphatidylinositol 3-kinase (PI3K) and protein kinase B (AKT). Another branch accepts signals, such as platelet-derived growth factor, nerve growth factor, and epidermal growth factor, then communicates the information via Ras, mitogen-activated protein kinase kinase (MEK), and extracellular signal-related kinase (ERK). The other branches play roles as sensors of the cellular energy status and the availability of amino acids. In the midstream area, these branches merge into a single flow at the TSC1 (hamartin)/TSC2 (tuberin) complex, a negative regulator of the system that inhibits the activities of Ras homolog enriched in brain (Rheb) and mTOR complex 1 (mTORC1) [2,3,4,5,6]. mTORC1 is a target molecule for pharmacological treatment with rapamycin and its derivatives (rapalogs). Downstream of mTORC1, the flow of signal transduction is divided again into multiple branches. One stream promotes protein synthesis via ribosomal protein S6 kinase (S6K) and ribosomal protein S6 (S6) [7], one enhances cap-dependent translation via eukaryotic translation initiation factor-4E (eIF4E)-binding proteins (4EBPs) and eIF4E [8,9,10], and another inhibits autophagy via ULK-51-like kinase 1 (ULK1) [11,12,13]. The mTOR system regulates various cellular functions, such as growth, proliferation, metabolism, and survival/death. In systemic organs, it is critically involved in multiple processes, including neurogenesis [14], nutrition [15], and immunity [16]. In the brain, its roles are essential in cerebral cortical development, synaptic functions, and brain activities, such as learning, cognition, and social functions [17,18,19].
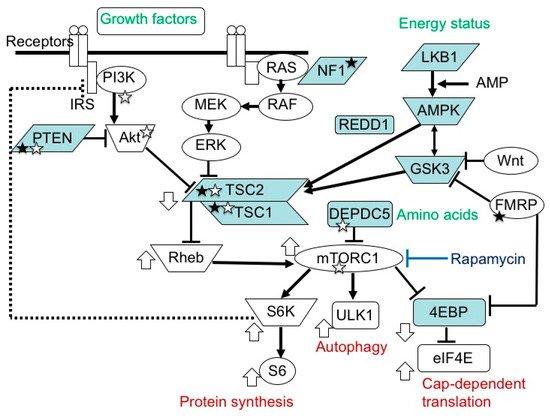
Figure 1. mTOR signaling pathway. In the upstream region, there are two paths, one transmitting signals of growth factors, and another transmitting those of energy status. In the midstream region, they converge at the TSC1/TSC2 complex into one main stream, which divides again at mTORC1 into S6K/S6 (protein synthesis), ULK1 (autophagy), and 4EBP/eIF4E (cap-dependent translation) pathways. The white circles and blue boxes represent factors that activate and inhibit, respectively, the activity of this system. Genetic defects in factors with a white and black star cause epilepsy and ASD, respectively.
Medical genetic studies have found many congenital disorders caused by a genetic defect in factors of the mTOR system, collectively referred to as mTORopathies. These conditions share the common brain symptoms of cerebral cortical dysgenesis, epilepsy, intellectual disability (ID), and/or autism spectrum disorder (ASD) [19]. Tuberous sclerosis complex (TSC) is a typical mTORopathy inherited in an autosomal dominant fashion. The two causative genes of TSC, TSC1 (chromosome 9q34) and TSC2 (16p13.3) [20,21], are located at the crossroad midstream of the mTOR pathway. Most brain symptoms of TSC are manifested by the dysregulation (hyperactivation) of the mTOR system. Since the 1990s, research has rapidly progressed from the identification of the genetic etiology [20,21] and elucidation of the molecular pathogenesis [22,23,24], to the development of molecular target therapies [25,26]. Currently, mTOR inhibitors are widely used in clinical practice to treat patients with TSC. Notably, they are efficacious not only for TSC-associated tumors, but also for some of the TSC brain symptoms, such as epilepsy [27].
2. Pathology and Clinical Picture of TSC
2.1. Systemic Findings
Recent advances in medical imaging and genetics have widened the clinical spectrum of TSC far beyond the classical triad of facial angiofibroma, epilepsy, and ID [28]. From a pathologic point of view, TSC is characterized by the multifocal occurrence of benign tumors (hamartomas) and focal dysplastic lesions (hamartias) in various organs, such as the skin, brain, eye, heart, lungs, and kidneys [29] (Table 1). In the vast majority of organs, these morphologic lesions, especially tumors, are the sole cause of functional problems, such as dysmorphism, rupture, and pressure to the surrounding normal tissues. In this context, the cerebrum is a remarkable exception as many patients with TSC have brain dysfunction, such as ID and ASD [30,31], without an apparent causal relationship with anatomical lesions. Thus, the clinical findings of TSC can be classified into three groups: (1) hamartoma, (2) focal dysplasia, and (3) brain dysfunction.
Table 1. Characteristic lesions of TSC (modified from Gomez (1999) [29]).
| Organ | Hamartias | Hamartomas |
|---|---|---|
| Skin | Hypomelanotic macules Confetti skin lesions |
Facial angiofibromas Fibrous cephalic plaque Ungual fibromas Shagreen patch |
| Brain | Cortical tuber | SEGA Subependymal nodules |
| Eye | Retinal achromic patch | Retinal hamartoma |
| Mouth | Gingival fibromas | Dental enamel pits |
| Lung | Pulmonary LAM MMPH |
|
| Heart | Cardiac rhabdomyoma | |
| Arteries | Wall defects/Aneurysm | Renal AML |
| Kidney | Renal cysts | |
| Bone | Bone cysts |
With regard to the occurrence/distribution of lesions and the severity of brain dysfunction, these clinical symptoms are remarkably variable among patients, which is also true for familial cases with the same mutation. The genotype–phenotype correlation is reported to be small [32,33,34], and the reason for the inter-case variability remains largely unknown. Each lesion or symptom shows a time course that is clearly age dependent. For instance, cardiac rhabdomyoma arises in the fetal period, facial angiofibroma in childhood, and pulmonary lymphangioleiomyomatosis (LAM) in adulthood (Table 2). Thus, the management of TSC patients requires life-long follow up.
Table 2. Age-dependent changes of lesions and symptoms in TSC.
| Lesion/Symptom | Pathology | Age of Occurrence or Worsening | Department in Charge | Note |
|---|---|---|---|---|
| Cardiac rhabdomyoma | Tumor | Fetal–neonatal period | Pediatric cardiology | Spontaneous regression in infancy |
| Cortical tuber | Dysplasia | Fetal–neonatal period/Infancy | Pediatric neurology | Focus of epileptic seizures |
| Hypopigmented macule | Dysplasia | Fetal–neonatal period/Infancy | Dermatology | |
| Epilepsy | Brain dysfunction | Infancy/Childhood | Pediatric neurology | Intractable in many cases |
| ID/ASD | Brain dysfunction | Infancy/Childhood | Pediatric neurology/Psychiatry | |
| Retinal hamartoma | Tumor | Infancy/Childhood | Ophthalmology | |
| Renal cyst | Dysplasia | Infancy/Childhood | Pediatrics | |
| SEGA | Tumor | Childhood/Adolescence | Neurosurgery | Hydrocephalus, potentially fatal |
| Facial angiofibroma | Tumor | Childhood/Adolescence | Dermatology | |
| TAND | Brain dysfunction | Childhood/Adolescence | Pediatrics/Psychiatry | |
| Renal AML | Tumor | Childhood/Adolescence/Adulthood | Pediatrics/Urology | Hemorrhage, potentially fatal |
| Pulmonary LAM | Tumor | Adolescence/Adulthood | Pulmonology | Predominantly affecting women, potentially fatal |
2.2. Brain Symptoms
2.2.1. Epilepsy
Many (about 80%) TSC patients have epileptic seizures of variable types. Typical clinical pictures are West syndrome (infantile spasms) in infancy, and focal epilepsy, which may occur in any age group [30,31]. In the vast majority of patients, the epileptic focus is located in or adjacent to a cortical tuber (focal dysplasia of the cerebral cortex). Epilepsy is often resistant to antiepileptic drugs, requiring neurosurgical treatment in many cases.
2.2.2. ID and ASD
The level of intelligence is variable among patients, ranging from normal to profound ID. ID is present in more than half of patients, and ASD in about half. Even patients with normal intelligence have a variety of behavioral, cognitive, and psychosocial problems, which are collectively called TSC-associated neuropsychiatric disorders (TAND) [35].
2.2.3. Brain Tumor
Approximately 10% of patients have subependymal giant cell astrocytoma (SEGA), a benign tumor on the wall of the lateral ventricle. A large SEGA may cause hydrocephalus and clinical signs of increased intracranial pressure. Hydrocephaly is usually ascribed to the occlusion of the foramen of Monro, although the exact mechanism is still poorly understood. For the treatment of SEGA, the first choice is surgical resection of the tumor. Chemotherapy with an mTOR inhibitor, everolimus, is also efficacious, and has recently become another choice of therapy [25].
3. Etiology and Pathogenesis of TSC
3.1. TSC Gene Mutations and Their Consequences
TSC is caused by various loss-of-function mutations in the two genes, TSC1 and TSC2 [33,34]. No hotspot mutation has been reported. A genotype–phenotype relationship has been noted, but it is small. There is no qualitative difference between TSC1 and TSC2. However, TSC2 mutations tend to show more severe brain symptoms and a larger propensity to develop tumors than TSC1 mutations [32,33,34].
TSC1 and TSC2 encode for tumor suppressors, hamartin and tuberin, respectively. These proteins bind to form a complex and then stabilize each other [2,3]. TSC-associated tumors show a decreased expression in both [36,37]. The TSC1/TSC2 complex is located in the midstream of the mTOR pathway and negatively regulates the activity of the system [2,3,4,5,6] (Figure 1).
3.2. Germline and Somatic Mutations
In patients with TSC, all the somatic cells have a germline mutation (first hit) in one allele of either the TSC1 or TSC2 gene, which can cause haploinsufficiency of the TSC1/TSC2 complex. When an additional somatic mutation (second hit) occurs during mitosis, the function of the TSC1/TSC2 complex becomes null.
Previous studies have demonstrated that TSC-associated tumors (hamartomas) occur according to the two-hit hypothesis [22,23,43]. The second hit is typically a small deletion of either 9q34 (TSC1) or 16p13.3 (TSC2), causing loss of heterozygosity (LOH), the incidence of which is reportedly high in kidney tumors (renal angiomyolipoma) but low in brain tumors (SEGA).
The genetic mechanisms of TSC-associated focal dysplasia (hamartias) largely remain to be elucidated. In cerebral dysplastic lesions, namely cortical tubers, LOH has not been found [22,23,43,44]. Studies using laser capture microdissection have found point mutations (but not LOH) in abnormal giant cells (astrocyte-like balloon cells and cytomegalic neurons), which are a histopathological hallmark of cortical tubers [24,45]. A cortical tuber comprises a small number of abnormal giant cells with null TSC1/TSC2 function, and a large number of normal-sized neurons/glial cells with haploinsufficiency [46]. The interaction of both cell types may account for the epileptogenicity of cortical tubers (Figure 2).
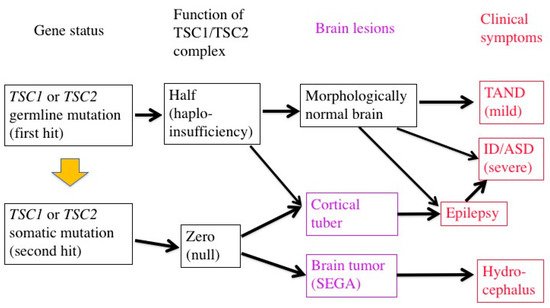
Figure 2. Two-hit hypothesis in a brain with TSC. The first hit, or germline mutation, can cause, by itself, mild TAND symptoms without any morphologic abnormalities. The second hit, or somatic mutation, may produce morphologic lesions (purple), either hamartoma (SEGA) or hamartia (cortical tuber). A tuber consists of a large number of “haploinsufficient” neurons and glial cells of normal size, and a small number of “null” cells of abnormal size and shape, and may become the focus of epileptic discharge. Severe early-onset epilepsy distorts the development of the brain network, leading to severe ID and/or ASD.
mTOR inhibitors are useful in the treatment of TSC-associated tumors in the brain, heart, and kidneys (everolimus) [25,26,47], as well as in the skin and lungs (sirolimus) [48,49]. Furthermore, clinical studies have recently shown that mTOR inhibitors are effective not only for tumors, phenotypes predominantly resulting from the second hit, but also for brain dysfunction (epilepsy) caused by a combination of the first and second hits [27]. The molecular basis of the efficacy is that the main stream of the mTOR system is essentially single between the TSC1/TSC2 complex and mTORC1 (Figure 1).
4. Brain Dysfunction in TSC
4.1. ID, ASD, and Epilepsy
Most TSC patients have a variety of behavioral, cognitive, and/or psychiatric problems, collectively termed as TAND. ID and ASD are the most common, affecting about 80% and 40% of patients, respectively. Notably, the distribution of intelligence quotient (IQ) is bimodal, dividing patients into two groups: profound ID with IQ (less than 30), and normal/subnormal mentality with a slight reduction in average IQ (around 90) [50] (Figure 3). Epidemiologic studies have shown that early-onset epilepsy (represented by West syndrome) is much more common in the former group than in the latter [51,52].
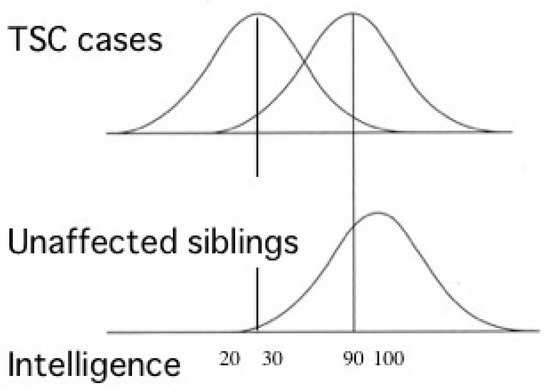
Figure 3. Cognitive function of patients with TSC showing bimodal distribution of IQ (modified from Joinson et al. (2003) [50]). Severe early-onset epilepsy is common in patients with profound ID (IQ < 30), but not in those with normal/subnormal intelligence (IQ around 90).
4.2. Pathophysiology: Neural and Glial Dysfunction
In TSC-associated epilepsy, the epileptic focus is usually located within or adjacent to a cortical tuber. As described above, a cortical tuber consists of both abnormal giant cells caused by a somatic mutation (second hit) and normal-sized neurons/glial cells with a germline mutation (first hit) only. The abnormal giant cells are abnormal, not only morphologically, but also functionally, as indicated by their immunohistochemical and electron microscopic features [56,57]. Pathologic collaboration (networking) between severely dysfunctional, abnormal giant cells and mildly dysfunctional, normal-sized neurons/glial cells forms the epileptogenic focus. Thus, in the brain, mild dysfunction due to germline mutations (first hit) can cause clinical symptoms, in sharp contrast to other organs where all the symptoms are due to somatic mutations (second hit). There is evidence for the dysfunction of autophagy/cell clearing, which is implicated in epileptogenesis [12,13,58,59].
Previous studies in human patients and animal models, either in vivo or in vitro, have documented various abnormalities of TSC neurons and/or glial cells (Figure 4). For instance, an astrocyte-specific conditional knockout of the Tsc1 gene impairs the astrocytic transport of glutamate [60]. The expression and function of gamma aminobutyric acid (GABA) receptors are abnormal in abnormal giant cells of cortical tubers [61,62,63,64,65]. Synapse formation is defective in human cortical tubers [66], as well as in cultured neurons of Tsc1 and Tsc2 knockout mice [67]. Synaptic pruning is also defective in Tsc2 knockout mice [68]. There are also abnormalities of synaptic functions, such as impaired long-term potentiation (LTP) and long-term depression (LTD) in the hippocampus of Tsc1/Tsc2 knockout mice [17,69,70]. These changes, in turn, cause an excitation/inhibition (E/I) imbalance toward hyperexcitation [71]. White matter also shows abnormal findings, which are implicated in the pathogenesis of ASD. Diffusion tensor imaging (magnetic resonance imaging) reveals an abnormal integrity of cerebral white matter, which is improved after pharmacologic treatment with an mTOR inhibitor [72,73]. Taken together, these changes in the morphology and function of neurons and glial cells lead to TSC-associated brain dysfunction: epilepsy, ID, and ASD [71].
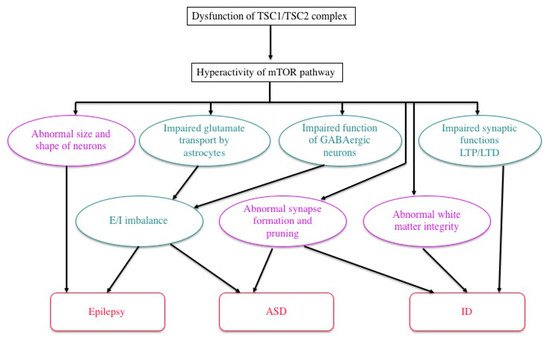
Figure 4. Possible mechanisms of TSC brain symptoms by which a loss of function of the TSC1/TSC2 complex causes a variety of structural (purple) and functional (green) abnormalities, leading to epilepsy, ID, and/or ASD (modified from Napolioni et al. (2009) [71]).
4.3. Efficacy of mTOR Inhibitors: Animal Experiment
To date, numerous animal models of TSC have been developed and used. One model, Eker rat, occurred spontaneously, whereas all the other models were produced artificially by genetic engineering. For translational research on the treatment of TSC-associated brain functions, we and other investigators often used mice produced by conventional gene knockout, due to their excellent construct validity.
The Eker rat, harboring a germline Tsc2 mutation, was originally found as a model of the hereditary cancer, renal cell carcinoma [74,75,76]. Compared to human TSC, the brain phenotype of the Eker rat is much milder, with a rare occurrence of cortical tuber-like lesions (focal cortical dysplasia) [77], an absence of epileptic seizures occurring spontaneously, and only a mild deficit in social interaction. Experimental studies introducing a second hit stimulus to the developing brain revealed that irradiation, but not a carcinogen, can increase the incidence of abnormal giant cells [78,79]. When the developing brain was pharmacologically exposed to severe epilepsy, the rat displayed ASD-like social deficit behavior [80].
Except for several Tsc1+/− mouse models [81,82], all heterozygous knockout mice for either Tsc1 or Tsc2 mutants showed neither cortical tubers nor spontaneous epileptic seizures. Thus, most Tsc1+/− and Tsc2+/− mice are not suitable as models of TSC-associated epilepsy, or brain symptoms caused by a combination of the first hit (normal-sized neurons and glial cells) and the second hit (abnormal giant cells). However, some of them did have mild, but recognizable, deficits in cognition and behavior [83]. For example, a Tsc2+/− mouse showed deficits in some hippocampal functions, such as spatial learning and contextual discrimination. Importantly, these deficits can be reversed by pharmacological treatment with rapamycin [17]. In two other strains, Tsc1+/− and Tsc2+/−, both male and female adult mice showed a deficit in social interaction, an ASD-like phenotype, which again was successfully improved by rapamycin. This improvement in behavior was accompanied by the normalization of gene expression and protein phosphorylation of the mTOR pathway factors [18]. These studies demonstrated that cognitive and behavioral problems in TSC, namely TAND, have not only structural, but also functional aspects, and that the latter can be improved even in adulthood. They also established Tsc1+/− and Tsc2+/− mice as models of mild TAND in the absence of early-onset epilepsy and illustrated their value in translational research of therapies for TAND.
4.4. Efficacy of mTOR Inhibitors: Clinical Trials
Since the beginning of this century, pharmacological treatment with mTOR inhibitors has rapidly progressed, especially for TSC-associated hamartomas: everolimus for brain tumors (SEGA) [25] and renal tumors (AML) [26], and sirolimus (rapamycin) for skin tumors (facial angiofibroma, topical) [48] and pulmonary tumors (LAM) [49]. When treated with mTOR inhibitors, these tumors shrink over several weeks or months. If the treatment continues, the decreased size remains unchanged over years, whereas the tumors often (but not always) grow back again if the treatment is discontinued. A significant difference in the efficacy, or the degree of reduction in size, is noted among patients, or among different types of tumors. For example, brain and kidney tumors decrease in volume, at best, by 70–80%, but never disappear. At worst, the tumor size remains unchanged [25,26], which nevertheless is usually acceptable in clinical practice because the tumors are benign in nature. For some tumors, the reasons for the inter-case or inter-lesion differences have been elucidated. In the treatment of renal AMLs with oral everolimus, the reduction in size is larger in fat-poor tumors than in fat-rich tumors [84]. In the treatment of skin tumors with topical sirolimus, the clinical effect is more rapid and remarkable in facial angiofibromas (those rich in blood vessels and covered by thin skin) than in other types of skin tumors (those poor in blood vessels and/or covered by thick skin) [85].
During clinical trials of oral (systemic) administration mTOR inhibitors, researchers noted their ancillary efficacy on brain dysfunctions, such as a decrease in epileptic seizures and the improvement of ASD symptoms. A phase 3, international clinical trial on the efficacy of everolimus for focal epilepsy (named EXIST-3) was conducted, and successfully demonstrated the usefulness of everolimus in the treatment of TSC-associated epilepsy by showing a clinically meaningful reduction in seizure frequency [27]. On the other hand, the inter-case variability was even larger in epilepsy than in cerebral and renal tumors; epileptic seizures totally disappeared in the best case, whereas they increased by three times in the worst case. With regard to ASD, a Japanese sub-study of EXIST-3 investigated the effects of everolimus on ASD symptoms using the Pervasive Developmental Disorders Autism Society Japan Rating Scale (PARS). Similar to the situation with epilepsy, the effect varied considerably among cases. Of the 11 patients treated by everolimus, 4 showed improvement (a decrease in PARS score by 5 or more) and one worsened (an increase by 5 or more) [86]. With regard to both epilepsy and ASD, the reasons for the large inter-case variability remain unclear. The usefulness of mTOR inhibitors as a therapy for ASD still remains to be established.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22136677
This entry is offline, you can click here to edit this entry!