Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Clinical Neurology
Neurotransmission is the process by which a signal is conveyed between neurons via endogenous signaling molecules called neurotransmitters. Neurotransmitters released from the axon terminal of one neuron cross the synaptic cleft and bind to receptors on the dendrites of another neuron, which are then converted into electrical signals. Synapse, the junction between neurons, has a tripartite structure that consists of presynaptic and postsynaptic nerve terminals along with the intimate association of glial cells.
- melatonin
- neurotransmission
- Alzheimer’s disease
1. Introduction
It is well established that neurotransmission is affected in patients with AD, particularly the chemical neuroanatomy of monoaminergic systems including serotonergic and dopaminergic systems, cholinergic systems including acetylcholine and GABA, and glutamatergic systems [1]. Neurotransmission dysfunction mainly arises from the degeneration of neurons as a result of the toxic accumulation of Aβ plaques (see Figure 1 and Table 1).
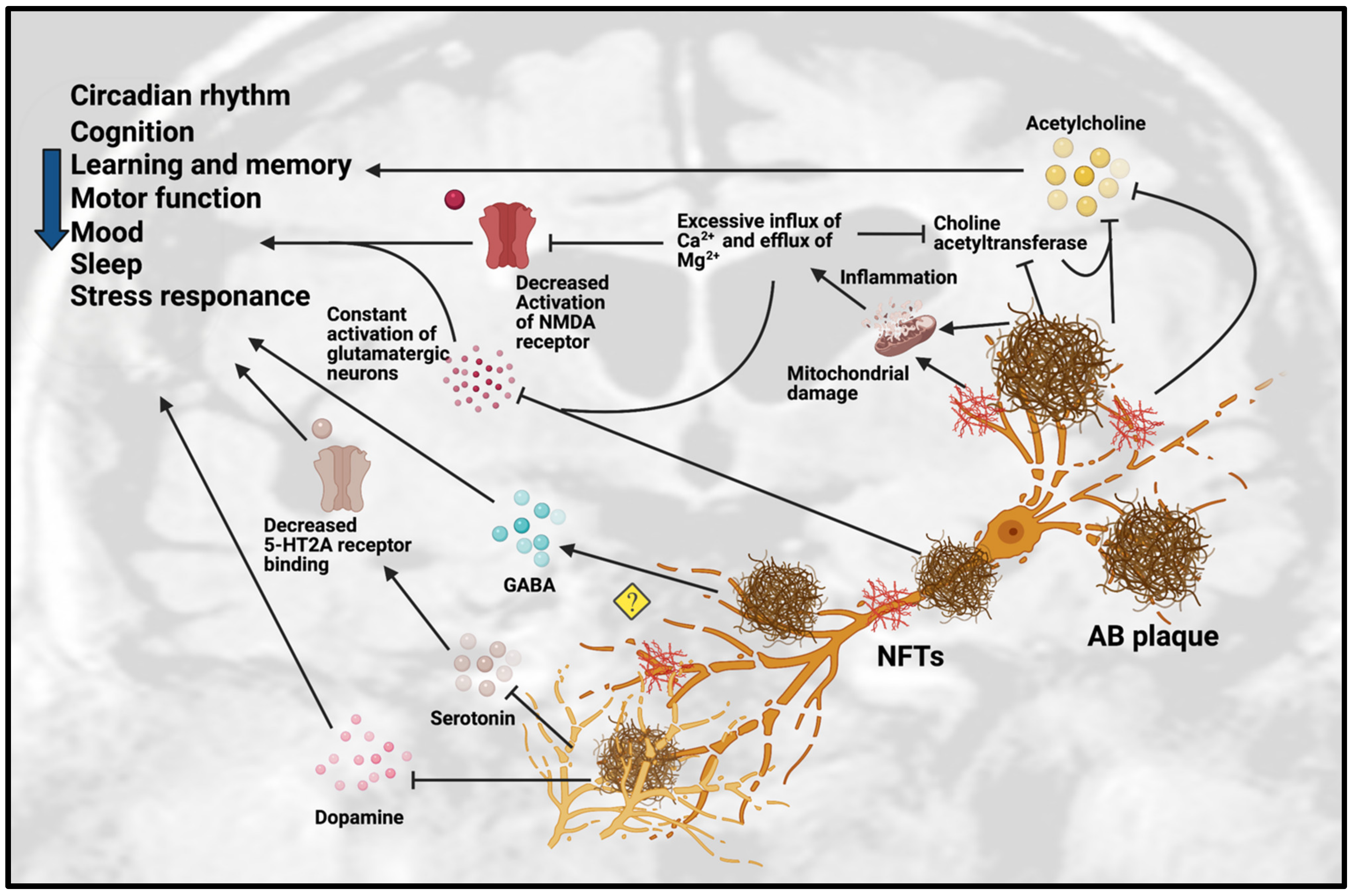 Figure 1. Dysfunction of neurotransmission in AD. Accumulation of Aβ plaques and NFT in AD cause impairment of the circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. These pathologies are toxic to neurotransmission systems, affecting cholinergic, glutamatergic, serotonergic and dopaminergic systems. Amyloid-beta plaques and NFT can inhibit the release of ACh and choline acetyltransferase, an enzyme that regulates ACh synthesis, which reinforces the inhibition effect of ACh. Amyloid-beta plaques and NFT can cause mitochondrial damage in glutamatergic neurons. The mitochondrial damage leads to inflammation due to excessive influx of Ca2+ and excessive efflux of Mg2+ that affect the activation of glutamatergic neurons and decreases the activation of NMDA receptor. The excessive influx of Ca2+ in glutamatergic neurons leads to inhibition of choline acetyltransferase and further inhibits the synthesis of ACh. However, the detailed mechanisms are not yet understood, as some studies showed the upregulation of GABA in certain regions but downregulation of GABA in other regions. Amyloid-beta plaques also disrupt the homeostatsis of serotonin (5-HT) by inhibiting the binding of serotonin receptor (5-HT2A) and disrupting the dopaminergic system. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.
Figure 1. Dysfunction of neurotransmission in AD. Accumulation of Aβ plaques and NFT in AD cause impairment of the circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. These pathologies are toxic to neurotransmission systems, affecting cholinergic, glutamatergic, serotonergic and dopaminergic systems. Amyloid-beta plaques and NFT can inhibit the release of ACh and choline acetyltransferase, an enzyme that regulates ACh synthesis, which reinforces the inhibition effect of ACh. Amyloid-beta plaques and NFT can cause mitochondrial damage in glutamatergic neurons. The mitochondrial damage leads to inflammation due to excessive influx of Ca2+ and excessive efflux of Mg2+ that affect the activation of glutamatergic neurons and decreases the activation of NMDA receptor. The excessive influx of Ca2+ in glutamatergic neurons leads to inhibition of choline acetyltransferase and further inhibits the synthesis of ACh. However, the detailed mechanisms are not yet understood, as some studies showed the upregulation of GABA in certain regions but downregulation of GABA in other regions. Amyloid-beta plaques also disrupt the homeostatsis of serotonin (5-HT) by inhibiting the binding of serotonin receptor (5-HT2A) and disrupting the dopaminergic system. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.Table 1. In vivo studies related to neurotransmission in AD. Abbreviations: 5 HT, 5-hydroxy-tryptamine, Serotonin; ACh, Acetylcholine; AChE, Acetylcholinesterase; APPswe, Amyloid-beta precursor protein with Swedish mutation; Aβ, Amyloid-beta; AβO, Amyloid-beta oligomers; AβPP, Amyloid-beta precursor protein; ChAT, Choline acetyltransferase; DA, Dopamine; DAergic, Dopaminergic; GABA, Gamma-aminobutyric acid; MAergic, Monoaminergic; mGlu2, Metabotropic glutamate receptor 2; NA, Noradrenergic; NMDAR, N-methyl-D-aspartate receptor; PS, presenilin transgenic; SN, Substantia nigra; TH-, Tyrosine hydroxylase negative; TH+, Tyrosine hydroxylase positive; VTA, Ventral tegmental area.
| Animal Model. | Gender | Age | Pathology Involved | Neurotransmission Dysfunction | Behavioral Effects | References |
|---|---|---|---|---|---|---|
| APPswe/PS1dE9 mice | N/A | 4–18 months old | Degeneration and loss of forebrain 5-HT and NA axons after Aβ deposits | Monoaminergic neurodegeneration | Anxiety-related behaviors in 18 months | [2] |
| Swiss mice treated with AβO | N/A | 3 months old | Development of Aβ plaques | AβO disrupts 5-HT homeostasis | Depressive-like behavior | [3] |
| APPswe/PS1dE9 mice | Male | 4, 8, 11 months old | Progressive accumulation of Aβ protein. | Significant decrease in 5-HT2A receptor binding | Memory impairment | [4] |
| 5xFAD mice | Male | 6 months old | Significant decrease of both TH+ and TH- cells in DA-producing areas | SN-VTA networks are enhanced to the synchronization of neuronal firing activity in DA-producing nuclei |
|
[5] |
| Tg2576 mice | Male | 2 and 6 months old | Degeneration of VTA DAergic neurons | Reduced noradrenergic transmission in dorsal subiculum | Age-related impairment of memory and non-cognitive functions | [6] |
| Tg2576 mice | N/A | 4–6 and 9–11 months old | Aβ were prominent in 20-month-old mice | Reduced ACh release from hippocampus in 9- to 11-month-old mice | Memory impairment present in 9- to 11-month-old mice | [7] |
| APP/PS1 mice | N/A | 3 and 7 months old | Aβ plaques deposition after cholinergic degeneration |
|
|
[8] |
| APP/PS1 and 5xFAD mice | N/A | 8 and 13 months old | Aβ plaques deposition and reactive astrocytes | Aberrant increase in GABA release from reactive astrocytes | Impaired learning and memory | [9] |
| AβPP/PS mice | Male | 2–4 months old | Abnormal glutamate release precedes cognitive decline | Significantly increased potassium-evoked glutamate release in CA1 | Cognitive decline | [10] |
| AβPPswe-PS1dE9 mice | N/A | 6 months old | Deposition of Aβ plaques |
|
Impairment of cognitive function and memory | [11] |
| TgAPP23 mice | Male and female | 24 months old | Deposition of Aβ plaques and cholinergic degeneration |
|
N/A | [12] |
| PS2APP mice | Female | 20 or 24 months old | Deposition of Aβ plaques | Significant reduction of glutamate level in frontal cortex | N/A | [13] |
| TgAPP23 mice | N/A | 7–8 months old | Dysfunction of cholinergic and monoaminergic systems |
|
N/A | [14] |
| PDAPP mice | Male and female | 4–6 months old | Deposition of Aβ plaques | Reduced basal and evoked ACh release from hippocampus | Hyper-locomotor function | [15] |
| 3xTg-AD mice | Male and female | 2–4, 13–15 and 18–20 months old | Aβ plaques deposition with cholinergic degeneration and alteration of neurotrophic factors |
|
N/A | [16] |
| hAPP-J20 mice | N/A | 6 months old | Altered synaptic plasticity and cognitive function | Significantly decreased phospho GluN2B levels and hippocampal LTP | Impaired learning and memory | [17] |
| TgCRND8 mice | N/A | 2 and 7 months old | Aβ plaques deposition, oxidative stress, reactive glial cells and neurodegeneration | Reduced ChAT-positive neurons and ACh levels. | Cognitive impairment | [18] |
| PS2APP mice | Male | 5, 9, 13 and 17 months old | Deposition of Aβ plaques | Significant loss of mGlu2 receptors in entorhinal cortex and lacunosum moleculare regions | N/A | [19] |
| PS2APP mice | Male | 3–4 months old | Altered synaptic plasticity | Aberrant GluN2B-NMDAR function | N/A | [20] |
| PDAPP mice | Male | 2, 4, 12 and 24 months old | Aβ plaques deposition with cholinergic degeneration |
|
N/A | [21] |
| 3xTg-AD mice | N/A | 9–23 months old | Deposition of Aβ plaques | Reduced ChAT and AChE-positive neurons | N/A | [22] |
| TgCRND8 mice | Male | 3 months old | Deposition of Aβ plaques and neuronal degeneration |
|
Cognitive impairment | [23] |
| TgCRND8 mice | Male and female | 2–3 and 12–13 months old | Deposition of Aβ plaques |
|
N/A | [24] |
| TgCRND8 mice | Male | 3 months old | Dysfunction of dopaminergic system |
|
Cognitive impairment | [25] |
2. Role of Melatonin on Neurotransmission
The effects of melatonin on neurotransmission primarily involve improvements in cholinergic and glutamatergic systems (see Figure 2). As mentioned previously, Aβ plaques can impair the function of glutamatergic neurons and cause excessive influx of calcium, which lead to overstimulation and unnecessary release of AChE, resulting in reduced choline acetyltransferase and ACh levels.
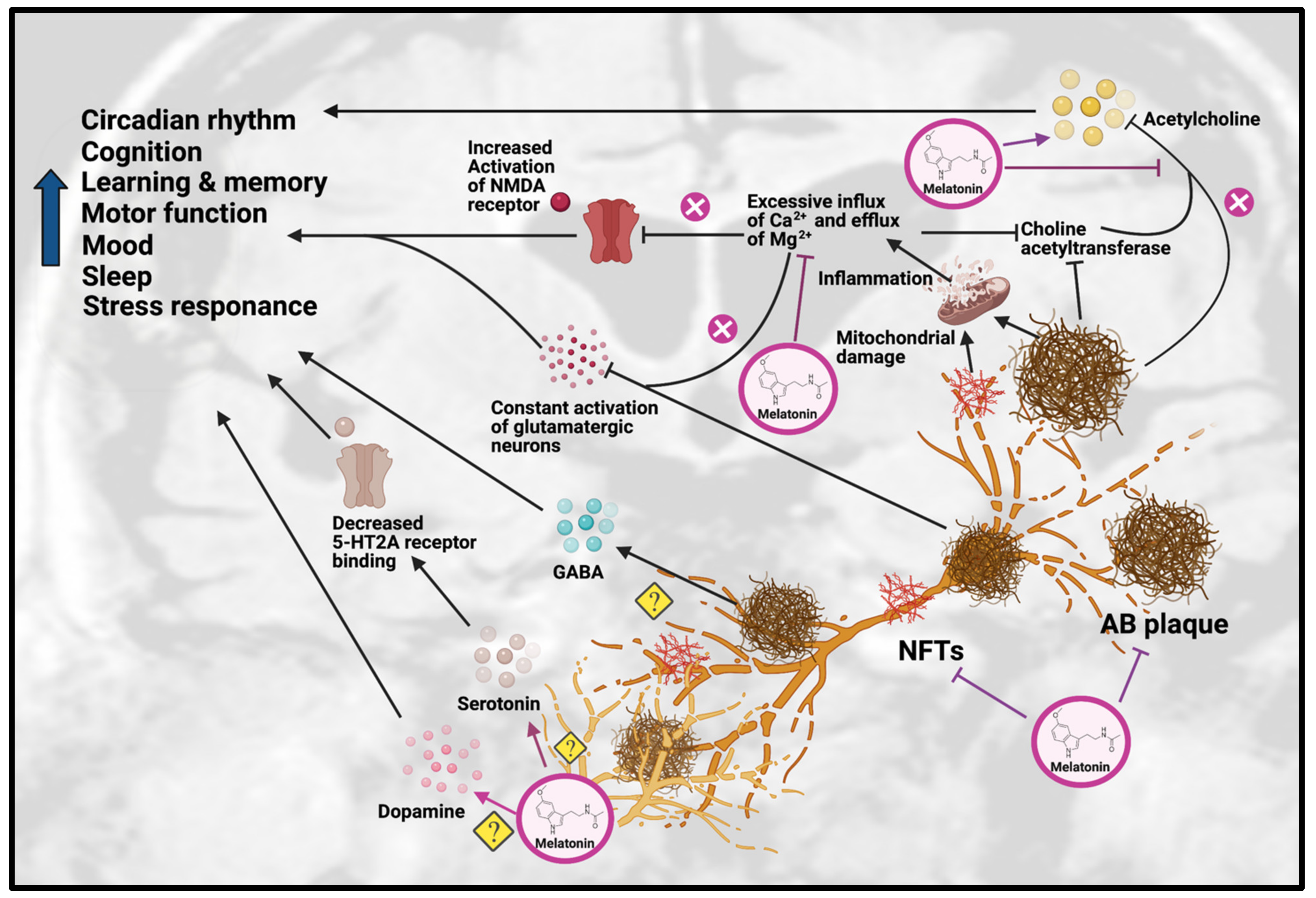 Figure 2. Effects of Melatonin treatment on dysfunction of neurotransmission in AD. Melatonin can ameliorate the formation of Aβ plaques and NFT, as well as improve the impairments due to these AD hallmarks, including disrupted circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. Melatonin treatment can have beneficial effects on serotonergic and dopaminergic systems, but the exact mechanisms have yet to be determined. Melatonin can also have beneficial effects on the cholinergic system by increasing acetylcholine release and reducing inflammation caused by excessive influx of Ca2+ and excessive efflux of Mg2+, thereby inhibiting choline acetyltransferase. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.
Figure 2. Effects of Melatonin treatment on dysfunction of neurotransmission in AD. Melatonin can ameliorate the formation of Aβ plaques and NFT, as well as improve the impairments due to these AD hallmarks, including disrupted circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. Melatonin treatment can have beneficial effects on serotonergic and dopaminergic systems, but the exact mechanisms have yet to be determined. Melatonin can also have beneficial effects on the cholinergic system by increasing acetylcholine release and reducing inflammation caused by excessive influx of Ca2+ and excessive efflux of Mg2+, thereby inhibiting choline acetyltransferase. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.Melatonin has been hypothesized to alleviate the disruption of the cholinergic system in AD through inhibiting the calcium-induced release of AChE, thus effectively acting as an acetylcholine enhancer [26]. Supporting this hypothesis, a study on a sporadic AD rat model showed that melatonin treatment could significantly decrease the level of inflammation and oxidation, as well as inhibit AChE activity [27]. In addition, it has been shown that as AD progresses, choline acetyltransferase [28] and AChE synthesis begins to decrease, which correlates positively with dementia severity in AD patients. Melatonin has also been shown to promote choline transport, which improved ACh synthesis [29]. In APP695 mice, melatonin treatment significantly decreased ChAT activity in the frontal cortex and hippocampus [30]. In a recent study on sporadic AD mice, melatonin rescued the AChE level and promoted neuroprotection [31].
Melatonin has also been suggested to alleviate the altered glutamatergic system in AD by inhibiting the activity of NMDA receptors. Melatonin was able to reduce excessive Ca2+ influx by altering the activity of voltage-gated Ca2+ channels, thereby inhibiting the effects of NMDA receptors [32]. This was supported by a study on adult male Wistar rats, which found that melatonin treatment attenuated the glutamatergic-dependent excitatory response in striatal neurons by reducing Ca2+ influx in voltage-gated Ca2+ channels and NMDA-gated Ca2+ channels, resulting in an anti-excitotoxic effect [33]. Even though these findings show melatonin has beneficial effects on cholinergic and glutamatergic systems, the impact of melatonin on the neurotransmission of monoaminergic systems has yet to be demonstrated, which will require more research.
3. Conclusions and Future Perspective
The effects of melatonin on neurotransmission and AD pathologies have been separately investigated in several respective studies. Melatonin is well established as a therapeutic for sleep disorders and jet lag, and has been investigated as an adjunct medication for cancer patients and as a medication for free-radical diseases. In this review, we have explored how melatonin can serve as a therapeutic for AD by inhibiting the pathological progression and restoring cholinergic and glutamatergic neurotransmission. Nevertheless, more research is needed to reveal its effects on other neurotransmitters such as GABA, serotonin, dopamine and histamine. Apart from the study on the direct interaction of melatonin with neurotransmission, the pathway through which melatonin can indirectly clear Aβ plaques is also worth studying since Aβ plaques are the fundamental source to cause neurotransmission dysfunction, and melatonin can reinforce the clearing effect of the glymphatic pathway by utilizing melatonin-AQP4 interaction. In addition, recent preclinical studies have indicated that melatonin metabolite N(1)-Acetyl-N(1)-formyl-5-methoxykynuramine, melatonin-derived benzylpyridinium bromides, melatonylvalpromide and melatonin-N,N-Dibenzyl(N-methyl) amine hybrids have neuroprotective effects against AD pathologies [34][35][36][37]. Moreover, melatonin receptor agonists piromelatine and agomelatine were found be effective against AD [38][39], but their neuroprotective effects against neurotransmission dysfunction in AD are still unknown. Further preclinical research is needed to investigate the detailed role of these compounds on different neurotransmission systems in AD, and further clinical studies on melatonin and its compounds are needed to validate their efficacy in the different stages of AD. To facilitate future studies, the recent advancement of neurotransmitter imaging techniques including positron emission tomography (PET) and Single-Photon Emission Computed Tomography (SPECT) can be taken into consideration. These are useful for accurate real-time neurotransmission detection. Furthermore, the improvement of the melatonin delivery system and the genetic variation of melatonin response can be evaluated by high-throughput screening and computer-aided drug design [40][41]. Finally, by understanding the mechanisms of how melatonin ameliorates AD pathogenesis, we can further ascertain its therapeutic value. Without a doubt, the future of melatonin as a potential treatment for AD is bright.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22136841
References
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.R.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Monoaminergic neuropathology in Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 101–138.
- Liu, Y.; Yoo, M.-J.; Savonenko, A.; Stirling, W.; Price, D.L.; Borchelt, D.R.; Mamounas, L.; Lyons, W.E.; Blue, M.E.; Lee, M.K. Amyloid Pathology Is Associated with Progressive Monoaminergic Neurodegeneration in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2008, 28, 13805–13814.
- Ledo, J.H.; Azevedo, E.P.; Beckman, D.; Ribeiro, F.C.; Santos, L.E.; Razolli, D.S.; Kincheski, G.C.; Melo, H.M.; Bellio, M.; Teixeira, A.L.; et al. Cross Talk Between Brain Innate Immunity and Serotonin Signaling Underlies Depressive-Like Behavior Induced by Alzheimer’s Amyloid- Oligomers in Mice. J. Neurosci. 2016, 36, 12106–12116.
- Holm, P.; Ettrup, A.; Klein, A.B.; Santini, M.A.; El-Sayed, M.; Elvang, A.B.; Stensbøl, T.B.; Mikkelsen, J.D.; Knudsen, G.M.; Aznar, S. Plaque Deposition Dependent Decrease in 5-HT2A Serotonin Receptor in AβPPswe/PS1dE9 Amyloid Overexpressing Mice. J. Alzheimers Dis. 2010, 20, 1201–1213.
- Vorobyov, V.; Bakharev, B.; Medvinskaya, N.; Nesterova, I.; Samokhin, A.; Deev, A.; Tatarnikova, O.; Ustyugov, A.A.; Sengpiel, F.; Bobkova, N. Loss of Midbrain Dopamine Neurons and Altered Apomorphine EEG Effects in the 5xFAD Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 70, 241–256.
- Cordella, A.; Krashia, P.; Nobili, A.; Pignataro, A.; La Barbera, L.; Viscomi, M.T.; Valzania, A.; Keller, F.; Ammassari-Teule, M.; Mercuri, N.B.; et al. Dopamine loss alters the hippocampus-nucleus accumbens synaptic transmission in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2018, 116, 142–154.
- Watanabe, T.; Yamagata, N.; Takasaki, K.; Sano, K.; Hayakawa, K.; Katsurabayashi, S.; Egashira, N.; Mishima, K.; Iwasaki, K.; Fujiwara, M. Decreased acetylcholine release is correlated to memory impairment in the Tg2576 transgenic mouse model of Alzheimer’s disease. Brain Res. 2008, 1249, 222–228.
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-Amyloid Deposition and Cognitive Impairment After Cholinergic Denervation in APP/PS1 Mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285.
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896.
- Hascup, K.N.; Hascup, E.R. Altered neurotransmission prior to cognitive decline in AβPP/PS1 mice, a model of Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 771–776.
- Tiwari, V.; Patel, A.B. Impaired glutamatergic and GABAergic function at early age in AβPPswe-PS1dE9 mice: Implications for Alzheimer’s disease. J. Alzheimers Dis. 2012, 28, 765–769.
- Boncristiano, S.; Calhoun, M.E.; Kelly, P.H.; Pfeifer, M.; Bondolfi, L.; Stalder, M.; Phinney, A.L.; Abramowski, D.; Sturchler-Pierrat, C.; Enz, A. Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J. Neurosci. 2002, 22, 3234–3243.
- von Kienlin, M.; Künnecke, B.; Metzger, F.; Steiner, G.; Richards, J.G.; Ozmen, L.; Jacobsen, H.; Loetscher, H. Altered metabolic profile in the frontal cortex of PS2APP transgenic mice, monitored throughout their life span. Neurobiol. Dis. 2005, 18, 32–39.
- Van Dam, D.; Marescau, B.; Engelborghs, S.; Cremers, T.; Mulder, J.; Staufenbiel, M.; De Deyn, P.P. Analysis of cholinergic markers, biogenic amines, and amino acids in the CNS of two APP overexpression mouse models. Neurochem. Int. 2005, 46, 409–422.
- Bales, K.R.; Tzavara, E.T.; Wu, S.; Wade, M.R.; Bymaster, F.P.; Paul, S.M.; Nomikos, G.G. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-Aβ antibody. J. Clin. Investig. 2006, 116, 825–832.
- Perez, S.E.; He, B.; Muhammad, N.; Oh, K.-J.; Fahnestock, M.; Ikonomovic, M.D.; Mufson, E.J. Cholinotrophic basal forebrain system alterations in 3xTg-AD transgenic mice. Neurobiol. Dis. 2011, 41, 338–352.
- Zhang, L.; Qin, Z.; Sharmin, F.; Lin, W.; Ricke, K.M.; Zasloff, M.; Stewart, A.F.; Chen, H.-H. Tyrosine phosphatase PTP1B impairs presynaptic NMDA receptor-mediated plasticity in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2021, 156, 105402.
- Bellucci, A.; Luccarini, I.; Scali, C.; Prosperi, C.; Giovannini, M.G.; Pepeu, G.; Casamenti, F. Cholinergic dysfunction, neuronal damage and axonal loss in TgCRND8 mice. Neurobiol. Dis. 2006, 23, 260–272.
- Richards, G.; Messer, J.; Faull, R.L.; Stadler, H.; Wichmann, J.; Huguenin, P.; Bohrmann, B.; Mutel, V. Altered distribution of mGlu2 receptors in β-amyloid-affected brain regions of Alzheimer cases and aged PS2APP mice. Brain Res. 2010, 1363, 180–190.
- Hanson, J.E.; Pare, J.-F.; Deng, L.; Smith, Y.; Zhou, Q. Altered GluN2B NMDA receptor function and synaptic plasticity during early pathology in the PS2APP mouse model of Alzheimer’s disease. Neurobiol. Dis. 2015, 74, 254–262.
- German, D.C.; Yazdani, U.; Speciale, S.G.; Pasbakhsh, P.; Games, D.; Liang, C.L. Cholinergic neuropathology in a mouse model of Alzheimer’s disease. J. Comp. Neurol. 2003, 462, 371–381.
- Robertson, R.T.; Baratta, J.; Yu, J.; LaFerla, F.M. Amyloid-β expression in retrosplenial cortex of triple transgenic mice: Relationship to cholinergic axonal afferents from medial septum. Neuroscience 2009, 164, 1334–1346.
- Steele, J.W.; Brautigam, H.; Short, J.A.; Sowa, A.; Shi, M.; Yadav, A.; Weaver, C.M.; Westaway, D.; Fraser, P.E.; St George-Hyslop, P.H. Early fear memory defects are associated with altered synaptic plasticity and molecular architecture in the TgCRND8 Alzheimer’s disease mouse model. J. Comp. Neurol. 2014, 522, 2319–2335.
- Salek, R.M.; Xia, J.; Innes, A.; Sweatman, B.C.; Adalbert, R.; Randle, S.; McGowan, E.; Emson, P.C.; Griffin, J.L. A metabolomic study of the CRND8 transgenic mouse model of Alzheimer’s disease. Neurochem. Int. 2010, 56, 937–947.
- Ambrée, O.; Richter, H.; Sachser, N.; Lewejohann, L.; Dere, E.; de Souza Silva, M.A.; Herring, A.; Keyvani, K.; Paulus, W.; Schäbitz, W.-R. Levodopa ameliorates learning and memory deficits in a murine model of Alzheimer’s disease. Neurobiol. Aging 2009, 30, 1192–1204.
- Mayuri, S.; Piyarat, G.; Parichart, B.; Russel, J.R.; Jutamaad, S. Mechanisms of Melatonin in Alleviating Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 1010–1031.
- Tyagi, E.; Agrawal, R.; Nath, C.; Shukla, R. Effect of melatonin on neuroinflammation and acetylcholinesterase activity induced by LPS in rat brain. Eur. J. Pharm. 2010, 640, 206–210.
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933.
- Rosales-Corral, S.A.; Acuña-Castroviejo, D.; Coto-Montes, A.; Boga, J.A.; Manchester, L.C.; Fuentes-Broto, L.; Korkmaz, A.; Ma, S.; Tan, D.-X.; Reiter, R.J. Alzheimer’s disease: Pathological mechanisms and the beneficial role of melatonin. J. Pineal Res. 2012, 52, 167–202.
- Feng, Z.; Chang, Y.; Cheng, Y.; Zhang, B.-L.; Qu, Z.-W.; Qin, C.; Zhang, J.-T. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer’s disease. J. Pineal Res. 2004, 37, 129–136.
- Labban, S.; Alghamdi, B.S.; Alshehri, F.S.; Kurdi, M. Effects of melatonin and resveratrol on recognition memory and passive avoidance performance in a mouse model of Alzheimer’s disease. Behav. Brain Res. 2021, 402, 113100.
- Shi, Y.; Fang, Y.-Y.; Wei, Y.-P.; Jiang, Q.; Zeng, P.; Tang, N.; Lu, Y.; Tian, Q. Melatonin in Synaptic Impairments of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 63, 911–926.
- Escames, G.; Macías, M.; León, J.; García, J.; Khaldy, H.; Martín, M.; Vives, F.; Acuña-Castroviejo, D. Calcium-Dependent Effects of Melatonin Inhibition of Glutamatergic Response in Rat Striatum. J. Neuroendocr. 2001, 13, 459–466.
- Rong, K.; Zheng, H.; Yang, R.; Liu, X.; Li, L.; Chen, N.; Zhao, G.; Gong, C.; Deng, Y. Melatonin and its metabolite N (1)-acetyl-N (1)-formyl-5-methoxykynuramine improve learning and memory impairment related to Alzheimer’s disease in rats. J. Biochem. Mol. Toxicol. 2020, 34, e22430.
- Luo, X.-T.; Wang, C.-M.; Liu, Y.; Huang, Z.-G. New multifunctional melatonin-derived benzylpyridinium bromides with potent cholinergic, antioxidant, and neuroprotective properties as innovative drugs for Alzheimer’s disease. Eur. J. Med. Chem. 2015, 103, 302–311.
- Wang, X.-C.; Zhang, Y.-C.; Chatterjie, N.; Grundke-Iqbal, I.; Iqbal, K.; Wang, J.-Z. Effect of melatonin and melatonylvalpromide on β-amyloid and neurofilaments in N2a cells. Neurochem. Res. 2008, 33, 1138–1144.
- Buendia, I.; Egea, J.; Parada, E.; Navarro, E.; León, R.; Rodríguez-Franco, M.I.; López, M.G. The Melatonin–N, N-Dibenzyl (N-methyl) amine Hybrid ITH91/IQM157 Affords Neuroprotection in an in Vitro Alzheimer’s Model via Hemo-oxygenase-1 Induction. ACS Chem. Neurosci. 2015, 6, 288–296.
- He, P.; Ouyang, X.; Zhou, S.; Yin, W.; Tang, C.; Laudon, M.; Tian, S. A novel melatonin agonist Neu-P11 facilitates memory performance and improves cognitive impairment in a rat model of Alzheimer’disease. Horm. Behav. 2013, 64, 1–7.
- Yao, K.; Zhao, Y.-F.; Zu, H.-B. Melatonin receptor stimulation by agomelatine prevents Aβ-induced tau phosphorylation and oxidative damage in PC12 cells. Drug Des. Dev. Ther. 2019, 13, 387.
- Aldewachi, H.; Al-Zidan, R.N.; Conner, M.T.; Salman, M.M. High-Throughput Screening Platforms in the Discovery of Novel Drugs for Neurodegenerative Diseases. Bioengineering 2021, 8, 30.
- Salman, M.M.; Al-Obaidi, Z.; Kitchen, P.; Loreto, A.; Bill, R.M.; Wade-Martins, R. Advances in Applying Computer-Aided Drug Design for Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 4688.
This entry is offline, you can click here to edit this entry!